Independent work
2026
94. 1,4-Dithianes in Synthesis: Rapid Assembly of Indolizidine Alkaloid-Type Scaffolds Through Cationic Polyene Cyclization
Demeyere E., Vermeulen M., Bevernaege K, Winne J.M.
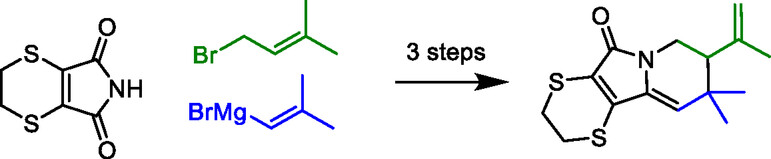
We report a synthetic approach to the indolizidine scaffold, fused to a 1,4-dithiane. In a straightforward three-step protocol, the commercially available building block 5,6-dihydro-1,4-dithiin-2,3-dicarboximide undergoes two consecutive alkenylations, followed by a cationic cyclization mediated by an α,β-unsaturated N-acyliminium ion, affording a small range of substituted indolizidine scaffolds. The reaction was originally observed as an unexpected side reaction, but offers an attractive entry into this class of biologically active alkaloids.
EurJOC, , p, 2026
https://doi.org/10.1002/ejoc.70703
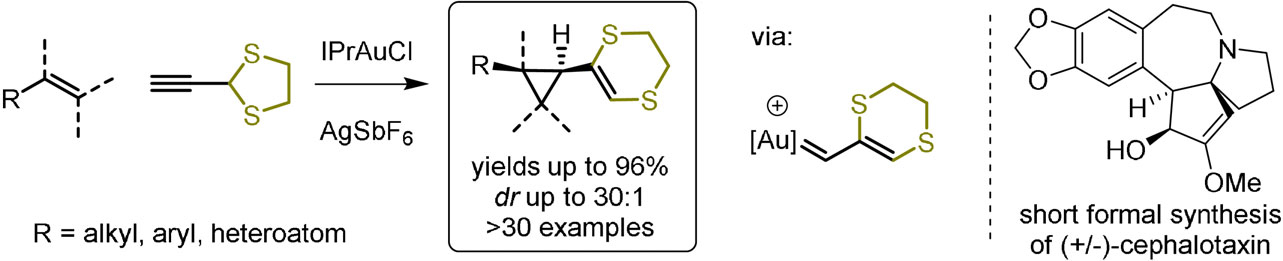
93. Stereoselective De Novo Synthesis of Substituted Cyclopentanes via a Gold(I)-Catalyzed (3 + 2) Cycloaddition of Enamides
Al-Khdhairawi A. A. Q., Yuan T., Van Hecke K., Jouffroy L., Winne J. M.
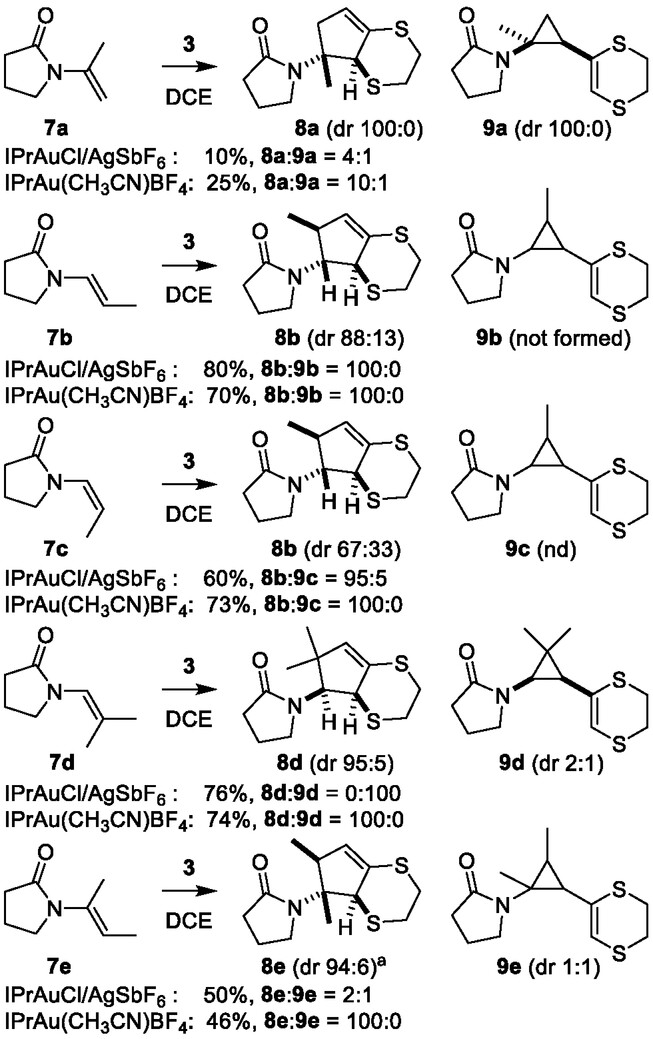
A general and stereoselective synthesis of highly substituted cyclopentylamine scaffolds is reported starting from various N-alkenyl-amides. The reaction involves the generation of an aurated dithioallyl cation which undergoes a classical stepwise (3 + 2) allyl cation cycloaddition with a range of enamide substrates. The close coordination of the reactive intermediates with the well-defined gold(I)-catalyst results in a reactivity and selectivity profile that can be readily rationalized on the basis of a nonconcerted carbocationic cycloaddition reaction, and some remarkable counterion effects are seen to effect the chemoselectivity of the overall process.
Advanced Synthesis & Catalysis, 368 (3), pe70182, 2026
https://doi.org/10.1002/adsc.70182
92. Chemical Upcycling of Polybutadiene Into Polyolefin-Based Dynamic Covalent Polymer Networks
Scholiers V., Vos C., Winne J. M., De Vos D., Du Prez F. E.
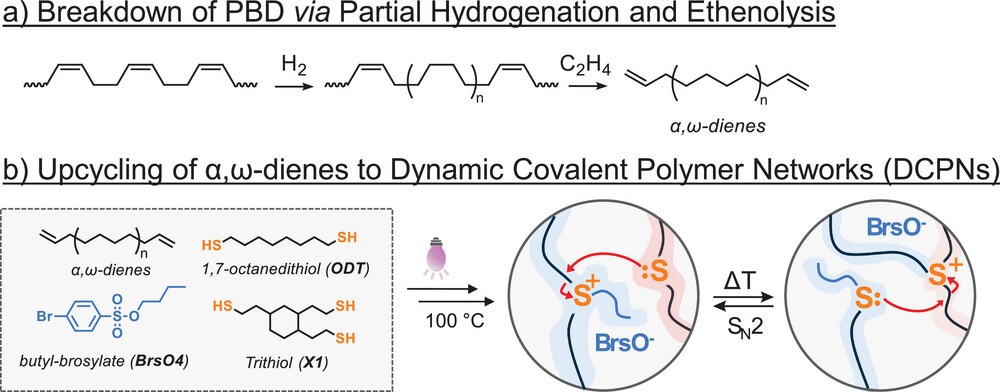
The use of non-renewable petrochemical feedstocks for the production of specialty polymer materials is not a sustainable practice. However, the use of waste commodity thermoplastic polymers as a feedstock for the production of specialty monomers is an attractive alternative. In this work, we show that high molar mass polybutadiene can be upcycled into reprocessable thermoset materials that can be recycled and reshaped multiple times as a dynamic covalent polymer network (DCPN). First, a one-pot partial hydrogenation and ethenolysis protocol was used to chemically cleave polybutadiene chains into low molecular weight α,ω-dienes. These were subsequently incorporated in a thiol-ene cured thermoset material using multifunctional thiols. The thioether linkages, connecting the polyolefin segments, can be turned into covalent dynamic moieties upon alkylation at elevated temperatures. The dynamic behavior of these thioether linkages was demonstrated through rheological and reprocessing experiments at elevated temperatures, highlighting their potential for sustainable material design. This strategy has a clear potential for an “upcycling” paradigm of bulk polymer waste into specialty polyolefin-based thermosets and DCPNs with appealing recyclability potential.
Macromolecular Rapid Communications, 47 (4), pe00818, 2026
https://doi.org/10.1002/marc.202500818
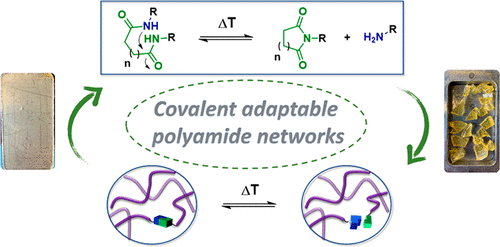
91. Creep Resistant and Reprocessable Polyamide Networks Based on Reversible Lactone Ring-Opening
Daelman B., Debuyck J., Scholiers V., Winne J. M., Du Prez F. E.
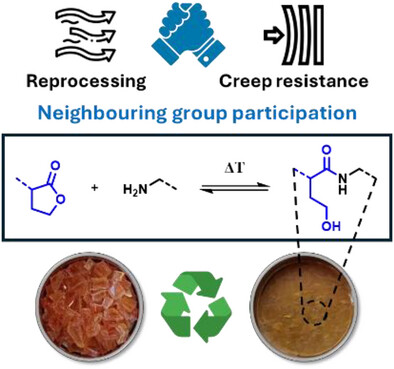
Amides are robust chemical linkages of interest in the field of dynamic covalent polymer networks (DCPNs), but their activation towards dynamic exchange remains an outstanding challenge. Herein, we introduce γ-hydroxy amides as a versatile motif for the design of creep resistant, yet fully reprocessable crosslinked polyamide-based materials. This was achieved by the reversible ring-opening of γ-lactones with primary amines. Small molecule kinetic studies showed that temperatures exceeding 120 °C are required for the ring-closure to occur at a significant rate. Thus, efficient exchange of the γ-hydroxy amide bonds was expected upon heating, while essentially non-dynamic covalent amide bonds should prevail at lower temperatures. γ-Hydroxy amides were then introduced into DCPNs, for which a newly prepared bifunctional γ-lactone monomer was cured with amine hardeners. A marked thermal response was observed in the rheological behavior, while creep resistance comparable to that of a non-dynamic epoxy-amine network was maintained up to 120 °C. Finally, we could also demonstrate the thermal resilience of γ-hydroxy amides after multiple compression molding cycles for a material with a glass transition temperature above 80 °C. Consequently, we expect that this simple chemistry platform has high potential for application in reprocessable thermoset materials.
Angewandte Chemie International Edition, 65 (5), pe19828, 2026
https://doi.org/10.1002/anie.202519828
2025
90. Exploring transamidation and chemical recycling of β-amino amide-derived covalent adaptable networks
Nguyen L. T., Tharayil A., Badi N., Winne J. M., Du Prez F. E.
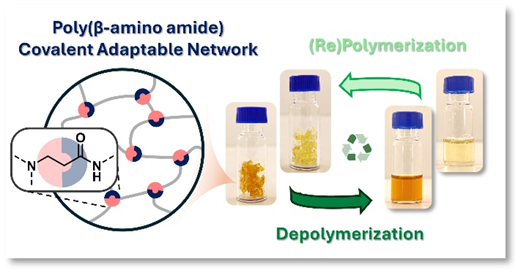
The growing environmental challenge of non-recyclable thermosets underscores the urgent need for sustainable alternatives. Covalent adaptable networks (CANs) containing dynamic β-amino amide moieties have emerged as promising reprocessable polymer networks, combining mechanical robustness with recyclability. In this work, we elucidate the exchange kinetics of both the well-established (retro)-aza-Michael addition and a newly identified transamidation pathway that is operative in β-amino amides. Systematic catalyst screening reveals that acidic catalysts significantly enhance viscoelastic control, thereby improving (re)processing efficiency. Furthermore, we introduce a chemical recycling protocol that enables the recovery of the original amino building blocks with up to 86% purity, demonstrating their direct use as feedstock in material (re)synthesis. These insights advance the fundamental understanding of dynamic bond exchange in β-amino amide based CANs and establish a viable route towards circular thermoset materials for numerous applications.
Polymer Chemistry, 16 (46), p5009–5018, 2025
https://doi.org/10.1039/d5py00959f
89. Dynamic β‑Amino Sulfonamides for the Synthesis of Covalent Adaptable Networks
Maes S., Nguyen L. T., Winne J. M., Du Prez F. E.
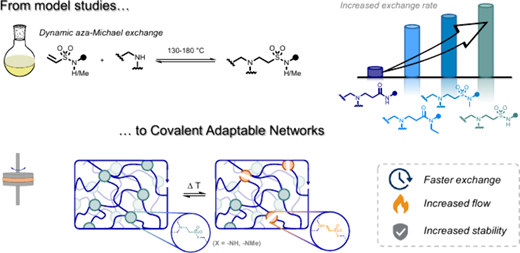
Thermosets are indispensable for their lightweight and excellent thermomechanical properties but suffer from nonrecyclability, exacerbating plastic pollution. Covalent Adaptable Networks (CANs), leveraging dynamic covalent bonds, address this challenge. This study explores β-amino sulfonamides (BASA) as novel dynamic and thermoreversible linkers for CANs. Compared to previously investigated β-amino amides (BAAs), small molecule BASA studies show a faster amine exchange reaction via (retro)aza-Michael reactions in a temperature window from 140 to 180 °C. More importantly, in contrast to the acrylamide monomers released from BAAs, the vinyl sulfonamide monomers released from BASAs are more robust and can resist homopolymerization at elevated temperatures. We showed that bisfunctional vinyl sulfonamide cross-linkers enable the synthesis of CANs with tunable properties, as verified through thermomechanical and rheological analysis. The anticipated increased thermal stability of BASA-based CANs was shown by the retention of mechanical and rheological performance over at least five recycling cycles.
ACS Macro Letters, 11 (14), p1721-1727, 2025
https://doi.org/10.1021/acsmacrolett.5c00658
88. Poly(styrene-co-maleamic acid)-based monoamide covalent adaptable networks
Hernandez A., Fischer S. M., Winne J. M., Du Prez F. E.

This study presents poly(styrene-co-maleamic acid)-based monoamide (PS-MMA) covalent adaptable networks (CANs) as a novel class of high performance dynamic covalent polymer networks. PS-MMA CANs are readily synthesized by crosslinking poly(styrene-co-maleic anhydride) (PSMA) copolymers with secondary diamines, introducing a previously unexplored dynamic monoamide exchange chemistry. By tailoring the amine-to-anhydride ratios, crosslink density and viscoelastic properties were finely adjusted, yielding networks with high thermal stability and reprocessability. The dissociation of monoamides into amines and anhydrides, as observed in high-temperature FT-IR analysis, was validated through Density Functional Theory (DFT) calculations. These calculations revealed an enthalpically favored tendency for amines and anhydrides to re-associate into monoamides, confirming their suitability for thermally triggered dynamics and effective viscosity control at increasing temperatures. Rheological analysis of the PS-MMA CANs showed distinct diamine structure-dependent profiles, where the interplay between the chain entanglements, supramolecular interactions and dynamic dissociative monoamide debonding governed their stress relaxation regimes and macroscopic flow behavior. Notably, such materials exhibited a unique combination of fast dissociative relaxation modes and slower reptation-driven dynamics, enabling precise control over material properties. These networks demonstrated an exceptional thermal resilience, maintaining their integrity and flow properties at temperatures up to 280 °C, surpassing the chemically analogous (and more widely studied) monoester-based CANs. Lastly, chemical recycling experiments further validated the sustainability of PS-MMAs, enabling efficient recovery of PSMA precursors while preserving their functionality.
Journal of meterials chemistry A, 13 (29), p24111-24125, 2025
https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta02035b
87. Thiol-thiol cross-clicking using bromo-ynone reagents
Nicque M., Meffert J. H., Maes D., Bevernaege K., Iftikhar M., Zwaenepoel O., Gettemans J., Madder A., Winne J. M.

Thiols are used in many click reactions, and are also excellent platforms for biomolecular click or bioconjugation reactions. The direct cross-coupling of two thiols is an attractive biomimetic concept for click chemistry, but leads to statistical mixtures of homo- and heterodimers. Here, we introduce a novel class of thiol-click reagents, bromo-ynones, where the kinetic differentiation between the first and second thiol addition onto these reagents facilitates a stepwise one-pot cross-clicking of two distinct thiols in aqueous media, without the need for intermediate isolation or purification. The two thiols are linked through a single carbon atom, mimicking a disulfide bridge. We demonstrate the use of bromo-ynones in the synthesis of various cross-coupled thiols, including small molecule drugs, fluorophores, carbohydrates, peptides and proteins, including an example of a protein-protein heterodimer. The resulting adducts are robust under physiological conditions and by judicious choice of the bromo-ynone reagent, the adducts can be stable even in the presence of excess free thiols.
Nature Communications, 16 (1), p, 2025
https://www.nature.com/articles/s41467-025-61682-5
86. Triazolinedione Modification of Prenylated Peptides, Proteins, and DNA: Toward a Single-Site Multilabeling Approach for Biomolecules
Tack L., Manicardi A., Leszczynska G., Dziergowska A., Ghielmetti A., Unal K., Decoene K., Winne J. M., Madder A.
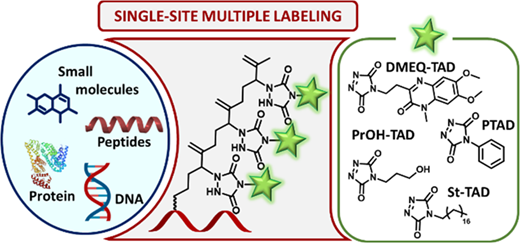
In efforts to shed light on the complexity of biological processes, attaching several or different payloads onto a biomolecular target of interest has become an interesting tool within the field of bioconjugation. Herein, we report on the exploitation of prenylated (bio)molecules in a 1,2,4-triazole-3,5(4H)-dione-based labeling strategy to develop a two-step single-site multiple labeling methodology that allows the introduction of up to three identical or different property-enhancing moieties. The methodology was first demonstrated on the small molecule targets farnesol and S-geranyl-2-thiouridine to be then applied to multi(functional) labeling of amino acids and peptides. We further demonstrate the usefulness of this approach to achieve branched lipidation of peptides, a strategy recently receiving more attention for enhanced cellular membrane localization and vaccine development. In addition, proof-of-concept experiments were performed involving single-site multiple labeling of large biomolecules such as farnesylated proteins and geranylated DNA.
ACS OMEGA, 10 (21), p21187-21194, 2025
https://doi.org/10.1021/acsomega.4c10125
85. Eliminating creep in vitrimers using temperature-resilient siloxane exchange chemistry and N-heterocyclic carbenes
Debsharma T., Nguyen L.T., Maliszewski B. P., Fischer S. M., Scholiers V., Winne J. M., Nolan S. P., Du Prez F. E.

This study explores a novel N-heterocyclic carbene-mediated siloxane exchange mechanism, laying the foundation for designing covalent adaptable networks (CANs) with high temperature stability (>200 °C) for dynamic covalent chemistry. Small molecule siloxane compounds, obtained by hydrosilylation reactions, are used to demonstrate siloxane-exchange via a mechanism supported by density functional theory. The proposed mechanism presents an equilibrium, at elevated temperatures, between an imidazolium salt and its free carbene form, which is the catalytically active species. Following this mechanistic insight, a tetra-substituted ester-terminated siloxane cross-linker was synthesized and cured with a commercial amine hardener. The ensuing ester–amine reaction yields thermally stable, non-dynamic amide bonds, thereby enhancing material stability. The resulting CANs exhibit rapid stress relaxation at elevated temperatures and demonstrate successful recycling through compression molding without any significant loss of material properties. Remarkably, the synthesized material showcases high creep resistance, even up to 150 °C, indicating the benefits of having a thermally reversible catalyst system for siloxane activation. This ground-up design of dynamic chemistry and material synthesis not only presents innovative material design but also suggests avenues for exploring thermally stable, fast-exchanging and yet creep-resistant CANs.
Chemical Science, 16 (21), p9337-9347, 2025
https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc06278g
84. Activated Phenyl Ester Vitrimers
Engelen S., Daelman B., Winne J. M., Du Prez F. E.
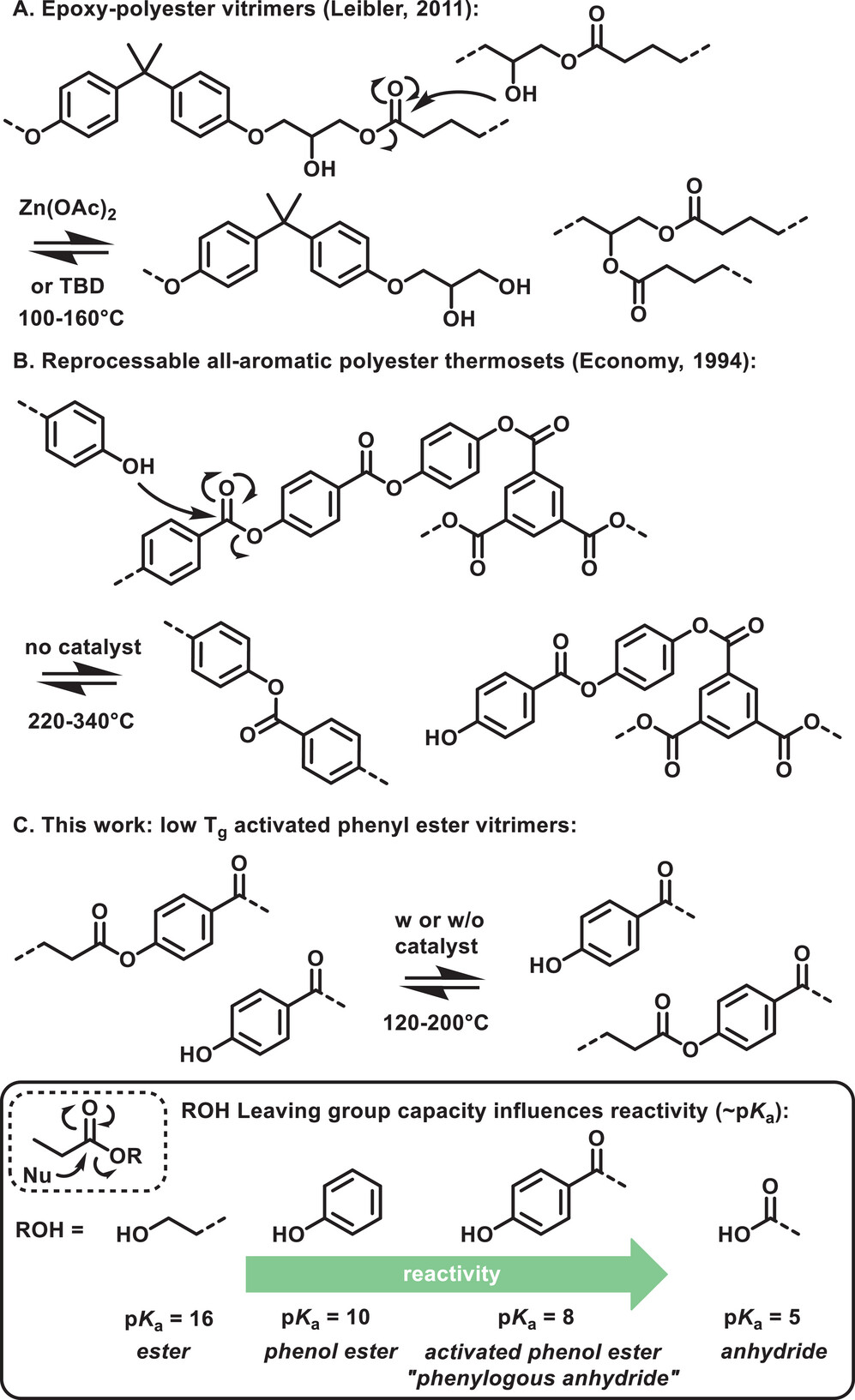
Aromatic esters are amongst the oldest known chemical motifs that allow for thermal (re)processing of thermosetting polymers. Moreover, phenyl esters are generally known as activated esters that do not require a catalyst to undergo acyl transfer reactions. Even though dynamic aromatic esters find applications in commercialized thermoset formulations, all-aromatic esters have found limited use so far in the design of covalent adaptable networks (CAN) as a result of their high glass transition temperature (Tg) and specific curing process. Here, a strategy to include partly aromatic esters as dynamic cross-links inside low Tg (-40 °C) thermosetting formulations, using aliphatic esters derived from para-hydroxybenzoic acid, which serves as a highly activated phenol or as a reactive phenylogous anhydride is reported. A small molecule study shows that the activated phenyl ester bonds can readily exchange with free phenol moieties at 200 °C under catalyst-free conditions, while the addition of a catalyst allows for a faster exchange. Robust and hydrophobic polymer networks are conveniently prepared via rapid thiol-ene UV-curing of unsaturated phenol esters. The obtained networks show high thermal stability (350 °C), fast processability, good water resistance, and low creep up to 120 °C, thus showing good promise as a platform for CAN.
Macromolecular Rapid Communications, 46 (4), p2400790, 2025
https://onlinelibrary.wiley.com/doi/10.1002/marc.202400790?af=R
83. Taking dynamic covalent chemistry out of the lab and into reprocessable industrial thermosets
Maes S., Badi N., Winne J. M., Du Prez F. E.
Dynamic covalent chemistry (DCC) allows the development of thermally (re)processable and recyclable polymer networks, which is a highly attractive feature for new generations of thermoset materials. However, despite a surge in academic interest wherein soon almost any imaginable DCC platform may have been applied in a thermoset formulation, dynamic or reversible covalent polymer networks have so far found only few industrial applications. This Review provides a perspective on the main strategies for the application of DCC in the design and development of bulk thermoset materials, and it presents some of the key hurdles for their industrial implementation. The polymer design strategies and associated chemistries are viewed from the perspective of how close to market their development pathway is, thus providing a roadmap to achieve high-volume breakthrough applications.
Nature Reviews Chemistry, 9 (3), p144-158, 2025
https://www.nature.com/articles/s41570-025-00686-7
82. Structural insights into brassinosteroid export mediated by the Arabidopsis ABC transporter ABCB1
Wei, H., Zhu H. Y., Ying W., Janssens H., Kvasnica M., Winne J. M., Gao Y. X., Friml J., Ma Q., Tan S. T., Liu X., Russinova E., Sun L. F.
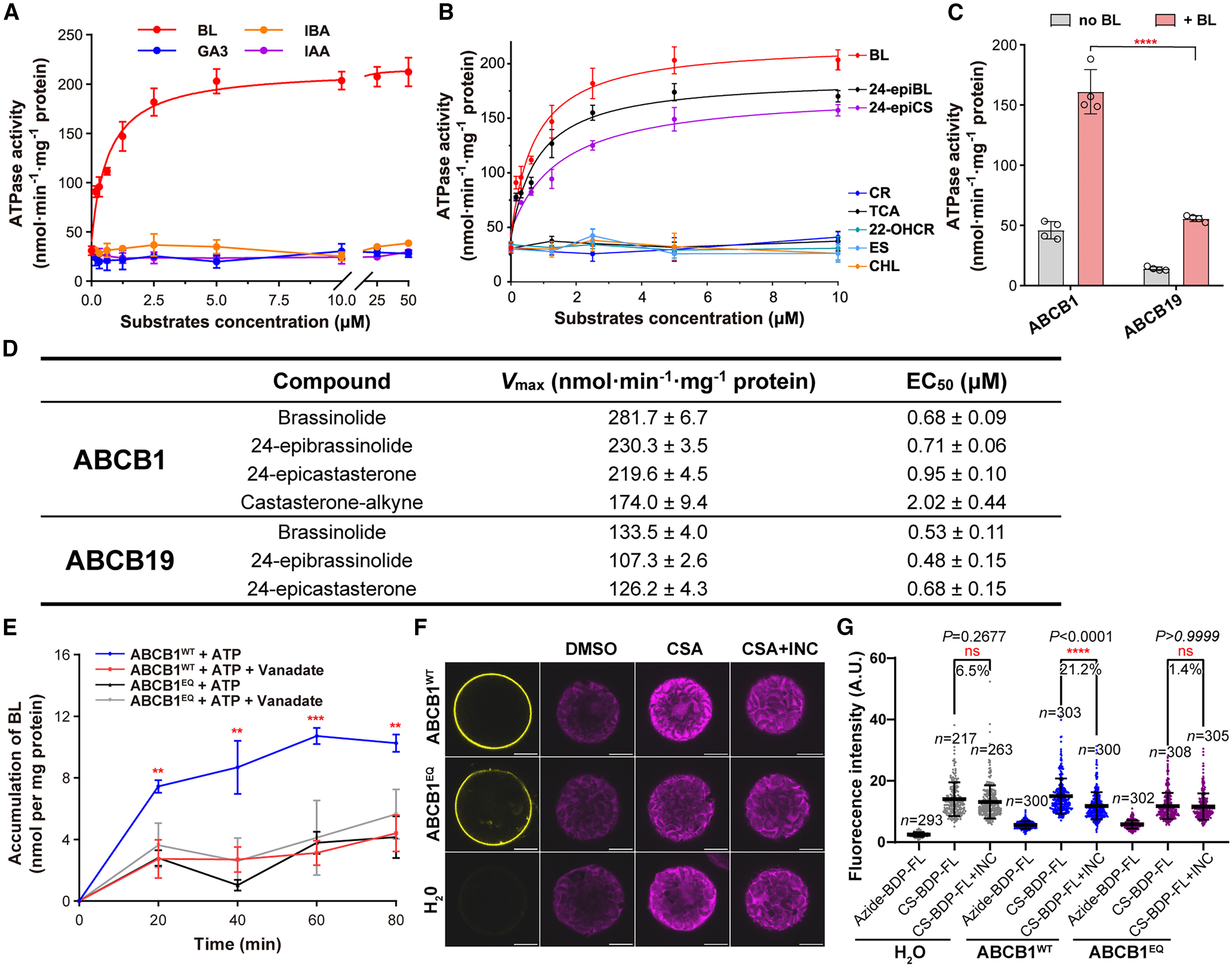
Brassinosteroids (BRs) are steroidal phytohormones indispensable for plant growth, development, and responses to environmental stresses. The export of bioactive BRs to the apoplast is essential for BR signaling initiation, which requires binding of a BR molecule to the extracellular domains of the plasma membrane-localized receptor complex. We have previously shown that the Arabidopsis thaliana ATP-binding cassette (ABC) transporter ABCB19 functions as a BR exporter and, together with its close homolog ABCB1, positively regulates BR signaling. Here, we demonstrate that ABCB1 is another BR transporter. The ATP hydrolysis activity of ABCB1 can be stimulated by bioactive BRs, and its transport activity was confirmed in proteoliposomes and protoplasts. Structures of ABCB1 were determined in substrate-unbound (apo), brassinolide (BL)-bound, and ATP plus BL-bound states. In the BL-bound structure, BL is bound to the hydrophobic cavity formed by the transmembrane domain and triggers local conformational changes. Together, our data provide additional insights into ABC transporter mediated BR export.
Plant Communications, 6 (1), p101181, 2025
https://www.sciencedirect.com/science/article/pii/S2590346224005972?via%3Dihub
2024
81. Tailoring the Reprocessability of Thiol-Ene Networks through Ring Size Effects
Scholiers V., Fischer S. M. Daelman B., Lehner S., Gaan S., Winne J. M., Du Prez F. E.
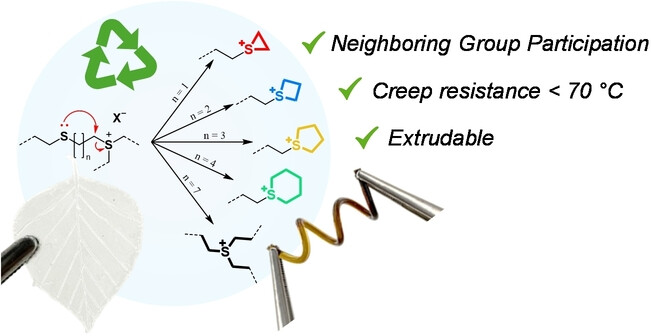
Recycling thermosetting materials presents itself as a major challenge in achieving sustainable material use. Dynamic covalent cross-linking of polymers has emerged as a viable solution that can combine the structural integrity of thermosetting materials with the (re-)processability of thermoplastics. Thioether linkages between polymer chains are quite common, and their use dates back to the vulcanization of rubbers. While it is known that thioether bonds can be triggered to exchange through transalkylation reactions, this process is usually slow, as thioether moieties not only have to be activated by an alkylating agent, but the activated thioether also has to associate with a second thioether moiety in a classical SN2-type process. Here, we present the rational design of dynamic polymer networks based on simple dithiol-based monomers and a fatty acid derived triene. Two neighboring thioethers can undergo a much faster bond exchange reaction, and we found that the exchange dynamics can be further tuned over almost three orders of magnitude by tailoring the distance between two thioether functionalities. This resulted in thioether-cross-linked materials that could be processed by extrusion, a continuous reprocessing technique that was previously not accessible for this class of cross-linked materials, while still exhibiting appealing creep-resistance below 70 °C
Angewandte Chemie International Edition, 64 (9), pe202420657, 2024
https://onlinelibrary.wiley.com/doi/10.1002/anie.202420657
80. SPL13 controls a root apical meristem phase change by triggering oriented cell divisions
Yang BJ., Sun YB., Minne M., Ge YH., Yue QR., Goossens V., Mor E., Callebaut B., Bevernaege K., Winne J. M., Audenaert D., De Rybel B.
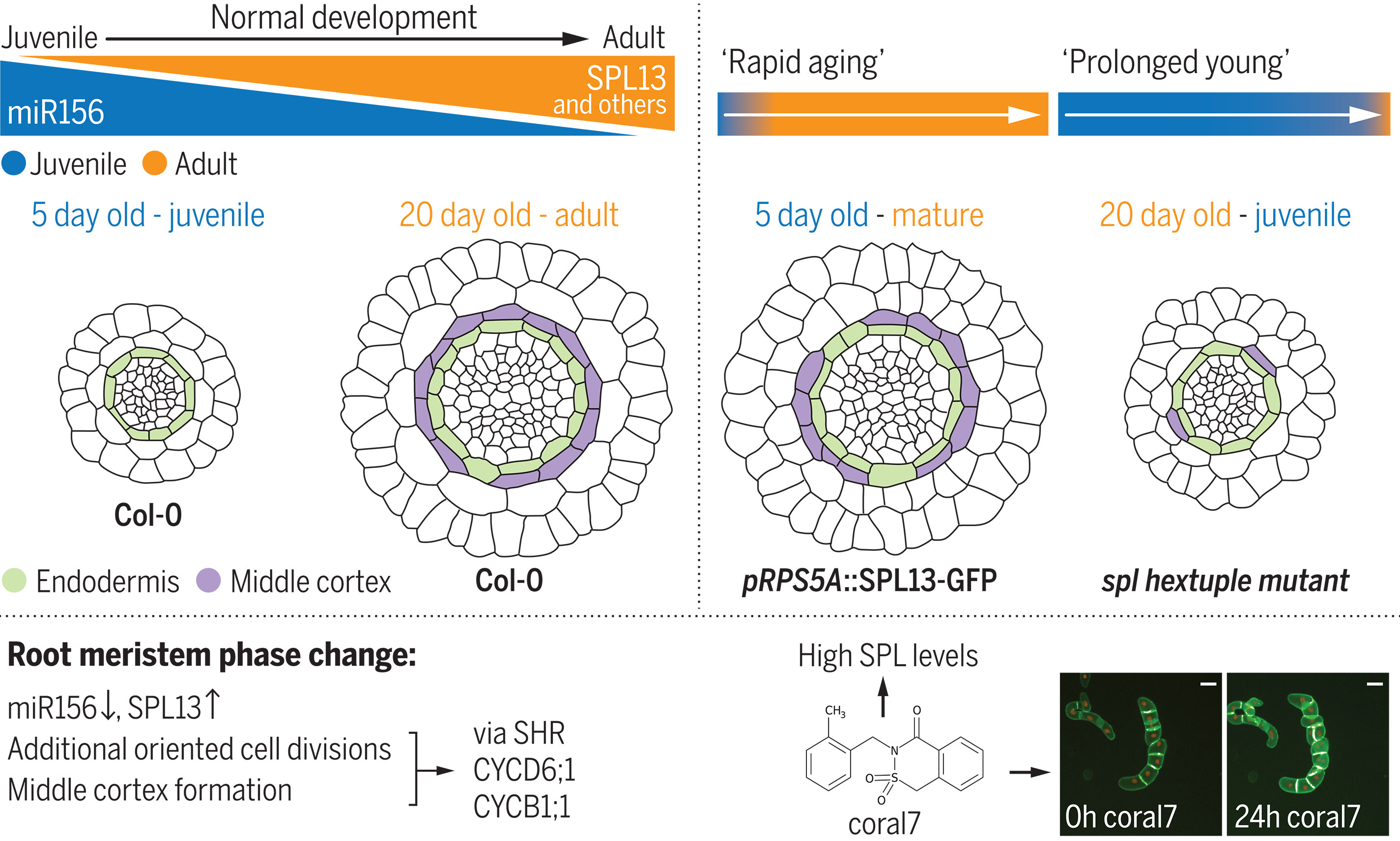
Oriented cell divisions are crucial for determining the overall morphology and size of plants, but what controls the onset and duration of this process remains largely unknown. Here, we identified a small molecule that activates root apical meristem (RAM) expression of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE13 (SPL13) a known player in the shoot's juvenile-to-adult transition. This expression leads to oriented cell divisions in the RAM through SHORT ROOT (SHR) and cell cycle regulators. We further show that the RAM has distinct juvenile and adult phases typed by morphological and molecular characteristics and that SPL factors are crucially required for this transition in Arabidopsis and rice (Oryza sativa). In summary, we provide molecular insights into the age-dependent morphological changes occurring in the RAM during phase change.
Science, 386 (6723), peado4298, 2024
https://www.science.org/doi/10.1126/science.ado4298
79. Triazolinedione-Based Cross-Linking of Plant Oils: An Introductory Organic Reactivity Laboratory Experiment
Maes D., Bevernaege K., Unal K., Denijs E., Winne J. M.
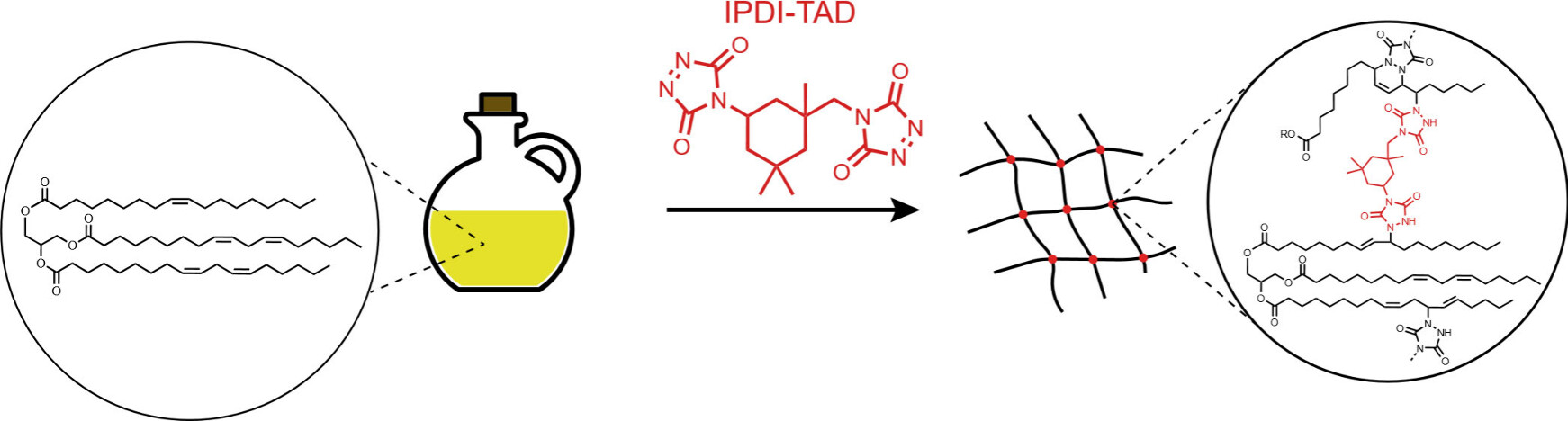
An undergraduate laboratory experiment has been developed for the synthesis of a reactive cross-linker that is useful in various highly visual demonstrations of organic reactivity, click chemistry, and biobased polymer synthesis. A triazolinedione-based cross-linker, which is known as a useful reagent for click chemistry, can be obtained from biobased isophorone diisocyanate (IPDI) in three synthetic steps: two steps to elaborate a urazole precursor, and finally a mild oxidation of this urazole to the highly reactive triazolinedione click reagent. As triazolinediones are strong chromophores, the progress of the oxidation step can be visually monitored by the appearance of a bright red-pink color. Students can perform either all three synthetic operations or just the final oxidation step, starting from a prepared batch of the precursor. Alternatively, for less experienced students, the reactive cross-linker or click reagent can be prepared beforehand by a lab assistant and used as such. Students used the freshly prepared click reagent to covalently cross-link various unsaturated plant oils into a polymer network. The gelation of the plant oil into a plant foil can be visually monitored, as the bright red-pink color of the cross-linker disappears over time. The purity of the synthesized cross-linking reagent can also be assessed by students by using a simple titration experiment. Students can rely on various practical first and second year undergraduate lab skills, but at the same time, they arrive at a tangible and visual experience of chemical reactivity.
Journal of Chemical Education, 101 (11), p4945-4951, 2024
https://pubs.acs.org/doi/10.1021/acs.jchemed.4c00279
78. Design and Continuous (Re)Processing of Thermally Resilient Poly(Styrene-co-Maleic Maleate)-Based Covalent Adaptable Networks
Hernandez A., Maiheu T., drockenmuller E., Montarnal D., Winne J. M., Du Prez F. E.
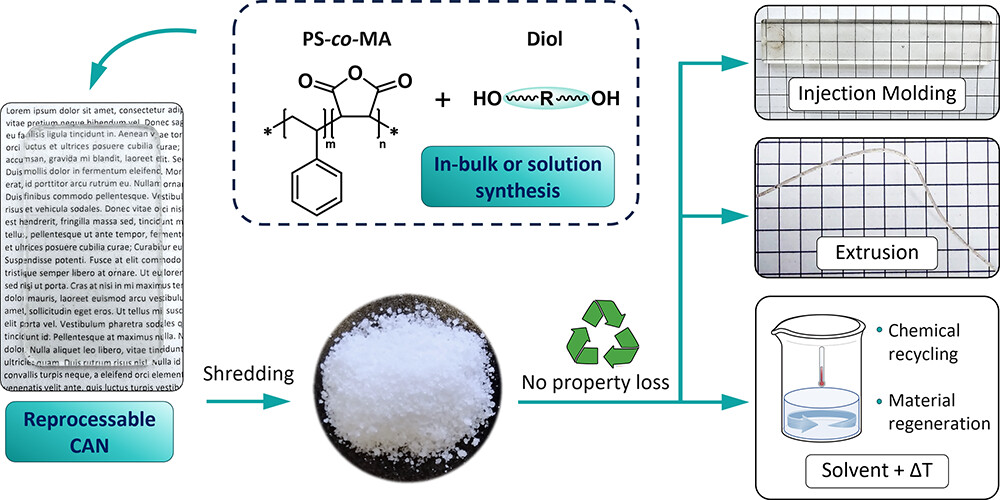
Cyclic anhydrides, classical monomers for bulk synthetic materials, have recently gained attention in covalent adaptable networks (CANs). They undergo reversible ring-opening with polyols, forming dynamic monoester linkages. This inherent property makes cyclic anhydrides ideal for industrially relevant polymer matrices with CAN-like recycling and reprocessing capabilities. Our study presents a rational design for poly(styrene-co-maleic maleate) monoester (PS-MME)-based polymer networks, achieved by cross-linking commercially available poly(styrene-co-maleic anhydride) (PS-co-MA) with a series of diols. The network synthesis is not only feasible on a large scale and under solvent-free conditions but also allows for the tailoring of the final cross-linking degree and properties by independently tuning the PS-co-MA maleic anhydride content, its chain length, and/or the loading of cross-linker diols used. Previous limitations in the thermal stability of MME-based CANs, stemming from remaining free carboxyl acids in monoester linkages, are circumvented through the dilution of reactive sites in an apolar matrix and switching to a cyclic anhydride ring embedded into the polymer backbone. This robust macromolecular architecture enhances the kinetics and thermodynamics of reversible monoester formation, resulting in PS-MMEs with exceptional thermal resilience. Even at temperatures exceeding 210 °C, PS-MMEs exhibit fast cross-link exchange without compromising their rheological, mechanical, or thermal properties. This feature not only enables continuous (re)processing via extrusion and injection molding but also allows the solvent-assisted recovery of the pure starting polyol monomers and PS-co-MA linear polymers, facilitating their chemical recycling and use in new polymer materials. In other words, this research paves the way for industrially applicable CANs by leveraging widely used bulk monomers and polymers.
Chemistry of Materials, 36 (15), p7487-7503, 2024
https://pubs.acs.org/doi/full/10.1021/acs.chemmater.4c01464
77. Dearomative (3 + 2) Cycloaddition of Indoles for the Stereoselective Assembly of Fully Functionalized Cyclopentanoids
Al-Khdhairawi A. A. Q., Yuan T., Van Hecke K., Winne J. M.
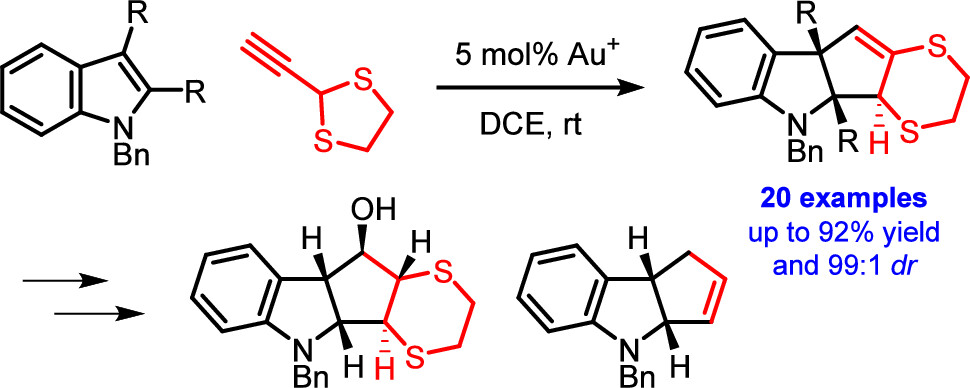
The gold(I)-catalyzed dearomative cyclopentannulation of various indoles with 2-ethynyl-1,3-dithiolane is reported. The method generates three new stereocenters with excellent control of relative stereochemistry and is tolerant of diverse functionalization and substitution patterns on the indoles. The obtained cyclopentane-fused indolines allow for interesting subsequent synthetic manipulations, giving rise to fully substituted cyclopentanes with control of the relative stereochemistry of all five stereocenters. The reported reaction illustrates and elucidates a mechanistic dichotomy underlying gold(I)-catalyzed reactions of 2-ethynyl-1,3-dithiolane.
Organic Letters, 26 (19), p4077-4081, 2024
https://doi.org/10.1021/acs.orglett.4c01139
76. Thermally Triggered Triazolinedione-Tyrosine Bioconjugation with Improved Chemo- and Site-Selectivity
Denijs E., Unal K., Bevernaege K., Kasmi S., De Geest B. G., Winne J. M.
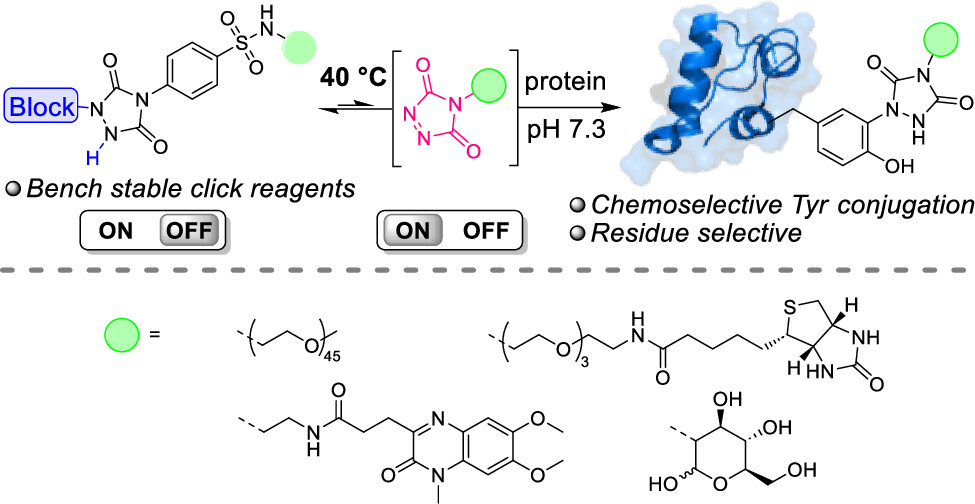
A bioconjugation strategy is reported that allows the derivatization of tyrosine side chains through triazolinedione-based "Y-clicking". Blocked triazolinedione reagents were developed that, in contrast to classical triazolinedione reagents, can be purified before use, can be stored for a long time, and allow functionalization with a wider range of cargoes and labels. These reagents are bench-stable at room temperature but steadily release highly reactive triazolinediones upon heating to 40 °C in buffered media at physiological pH, showing a sharp temperature response over the 0 to 40 °C range. This conceptually interesting strategy, which is complementary to existing photo- or electrochemical bioorthogonal bond-forming methods, not only avoids the classical synthesis and handling difficulties of these highly reactive click-like reagents but also markedly improves the selectivity profile of the tyrosine conjugation reaction itself. It avoids oxidative damage and "off-target" tryptophan labeling, and it even improves site-selectivity in discriminating between different tyrosine side chains on the same protein or different polypeptide chains. In this research article, we describe the stepwise development of these reagents, from their short and modular synthesis to small-molecule model bioconjugation studies and proof-of-principle bioorthogonal chemistry on peptides and proteins.
Journal of the American Chemical Society, 146 (18), p12672-12680, 2024
https://pubs.acs.org/doi/10.1021/jacs.4c02173
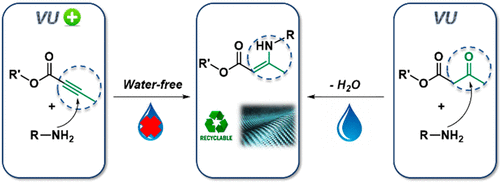
75. Thermoswitchable catalysis to inhibit and promote plastic flow in vitrimers
Van Lijsebetten F., Maes S., Winne J. M., Du Prez F. E.
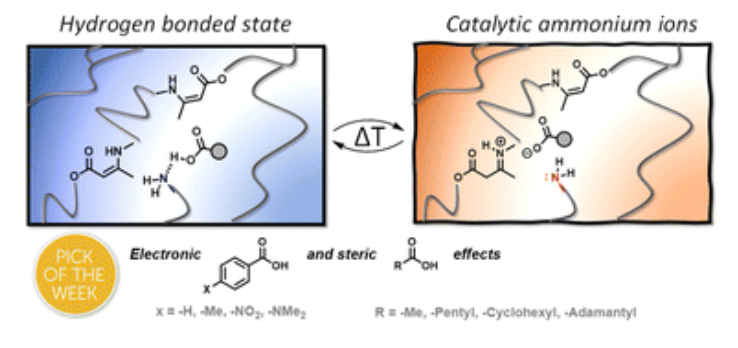
Acid-base catalysis is a common strategy to induce covalent bond exchanges in dynamic polymer networks. Strong acids or strong bases can promote rapid network rearrangements, and are simultaneously preferred catalysts for chemical reactions where maximum efficiency at the lowest possible temperature is aimed for. However, within the context of dynamic polymer networks, the incorporation of highly active catalysts can negatively affect the longer term application potential. Network dynamicity can diminish through catalyst ageing or quenching and highly active catalysts may prematurely activate bond exchanges, leading to dimensional instability and thus low creep resistance of the polymer networks. Herein, we present several examples where we explicitly explored weak acids (carboxylic acids) as catalysts for dynamic bond exchanges, using vinylogous urethanes (VU) as a well-understood protic acid catalysed vitrimer chemistry. Surprisingly, we have found that the sought-after long-term stability offered by a weak acid does not necessarily bring lower activity at high temperature. In fact, the weak acids show a remarkable thermoswitchable catalytic behaviour, going from an inactive hydrogen bonded state to an active state where the polymer matrix is protonated, with a profound impact on the network reactivity and rheology. Carboxylic acids with different electronic or steric environments show clear reactivity trends and their fine-tuning resulted in the most thermally responsive VU vitrimers studied to date. Our findings point out that catalyst choice and design for vitrimers is only poorly informed by catalyst performance in more traditional chemical reactions (in solvent), and that a more tailored catalyst design holds great promise for the field of vitrimers.
Chemical Science, 15 (19), p7061-7071, 2024
https://pubs.rsc.org/en/content/articlelanding/2024/sc/d4sc00417e
74. Structure and function of the Arabidopsis ABC transporter ABCB19 in brassinosteroid export
Ying W., Wang Y., Wei H., Luo Y., Ma Q., Zhu H., Janssens H., Vukašinovic N., Kvasnica M., Winne J. M., Gao Y., Tan S., Frimi J., Liu X., Russinova E., Sun L.
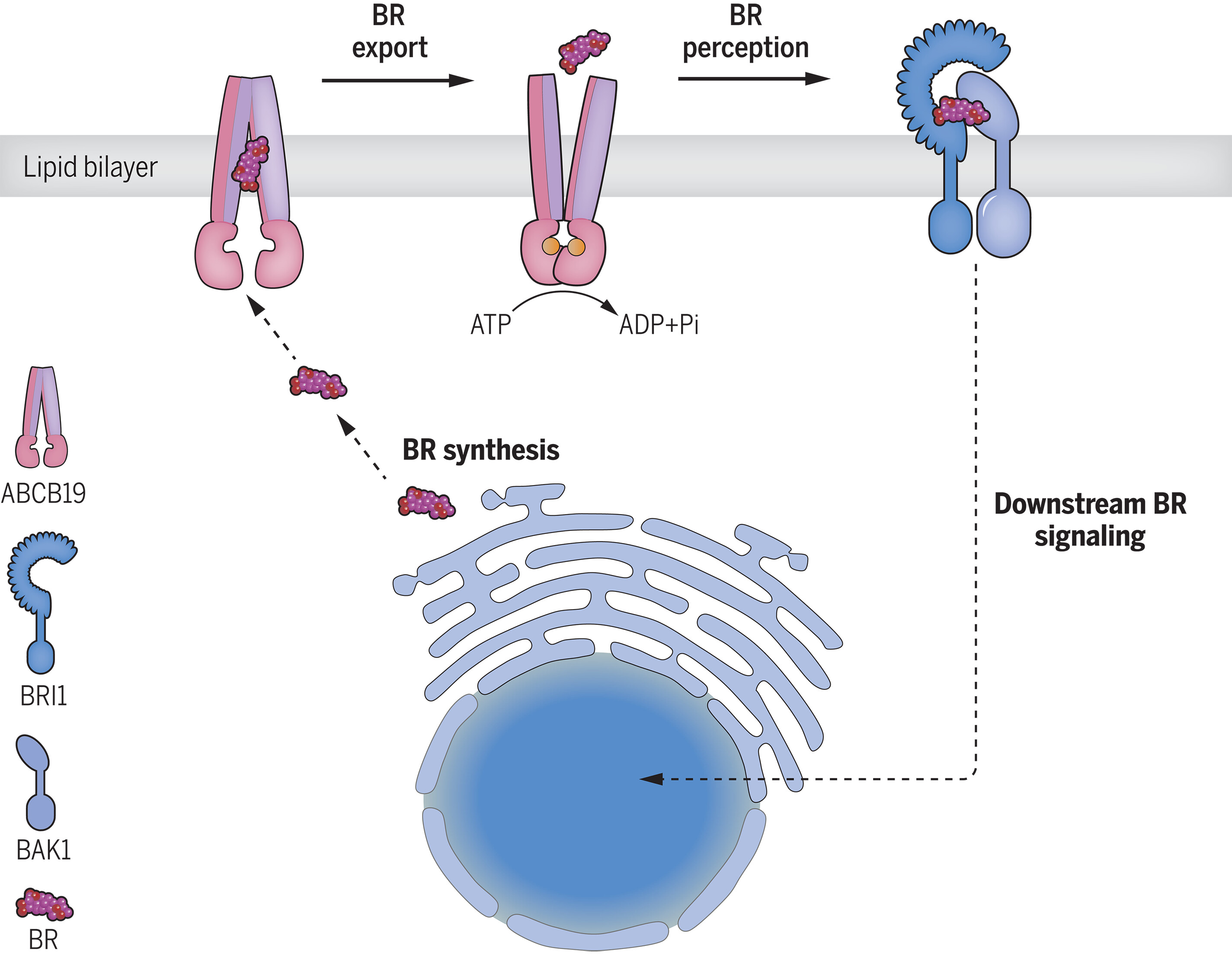
Brassinosteroids are steroidal phytohormones that regulate plant development and physiology, including adaptation to environmental stresses. Brassinosteroids are synthesized in the cell interior but bind receptors at the cell surface, necessitating a yet to be identified export mechanism. Here, we show that a member of the ATP-binding cassette (ABC) transporter superfamily, ABCB19, functions as a brassinosteroid exporter. We present its structure in both the substrate-unbound and the brassinosteroid-bound states. Bioactive brassinosteroids are potent activators of ABCB19 ATP hydrolysis activity, and transport assays showed that ABCB19 transports brassinosteroids. In Arabidopsis thaliana, ABCB19 and its close homolog, ABCB1, positively regulate brassinosteroid responses. Our results uncover an elusive export mechanism for bioactive brassinosteroids that is tightly coordinated with brassinosteroid signaling.
Science, 383 (6689), peadj4591, 2024
https://www.science.org/doi/10.1126/science.adj4591
73. Foam-to-Elastomer Recycling of Polyurethane Materials through Incorporation of Dynamic Covalent TAD-Indole Linkages
Unal K., Maes D., Stricker L., Lorenz K., Du Prez F. E., Imbernon L., Winne J. M.
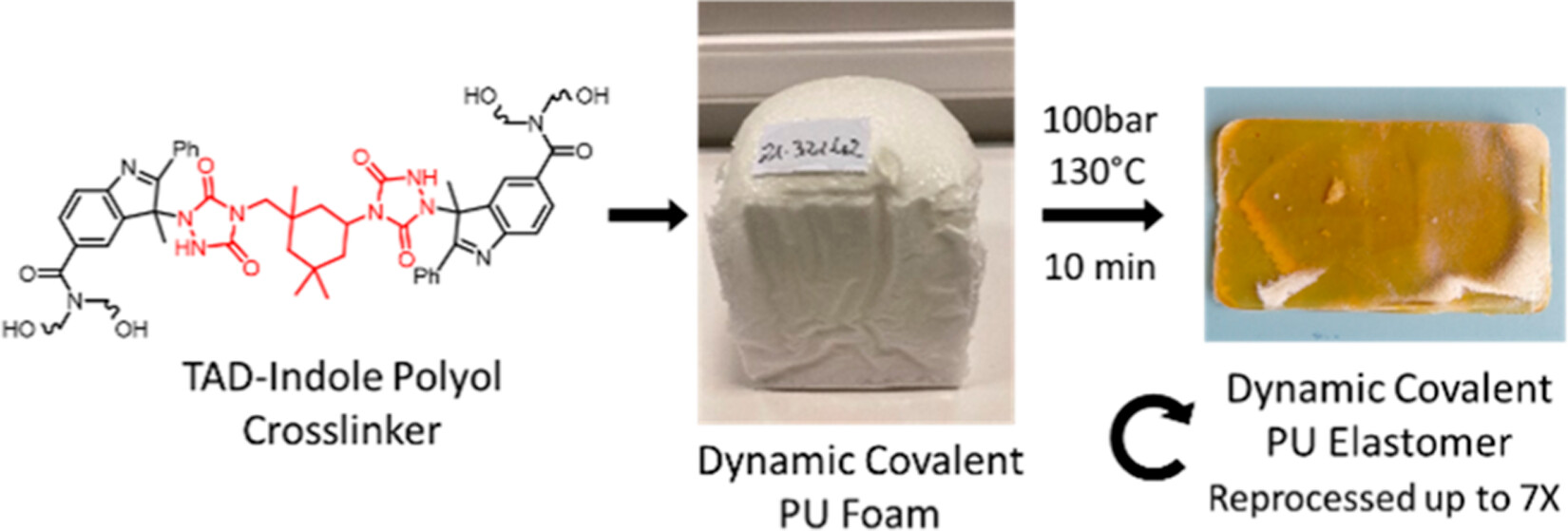
Polyurethane (PU) foams are a large volume commodity product and as such pose a considerable recycling challenge for the polymer manufacturing industry. The incorporation of dynamic covalent bonds within a PU network chemistry is a possible strategy to improve the sustainable development of PU foams. Herein, we report the outcome of a research program aimed at the incorporation of thermoreversible triazolinedione (TAD)-indole linkages within an industrial standard PU foam formulation. A scalable synthesis of the required TAD-indole building blocks was developed, aiming at maximizing their physicochemical compatibility with standard polyol and isocyanate PU foam ingredients. A pilot scale synthesis of a TAD-based cross-linker was developed, affording more than 50 kg of an IPDI-derived bis-urazole building block. Propoxylation of the indole fragments proved to be a key technology enabling the kilogram scale production of TAD-indole based polyols. The innovative dynamic covalent polyols were successfully used to produce a range of flexible PU foams and elastomers in a solvent-free foam formulation. Owing to the thermoreversible nature of the TAD-indole linkers, the PU foams could be processed into PU elastomers through thermal compression molding, which could be further recycled up to 7 times in the same manner. We studied the effect of network compositions on the foaming process and on the recycling efficiency. The thermal and mechanical properties of the materials have been studied with thermal analysis, tensile measurements, and rheology. This work demonstrates that TAD-indole chemistry is a viable strategy to improve polyurethane recycling but also points out some critical aspects and obstacles related to the design of such dynamic covalent PU foams.
Applied Polymer Materials, 6 (5), p2604-2615, 2024
https://doi.org/10.1021/acsapm.3c02791
72. Phenylpropynones as Selective Disulfide Rebridging Bioconjugation Reagents
Maes D., Nicque M., Iftikhar M., Winne J. M.
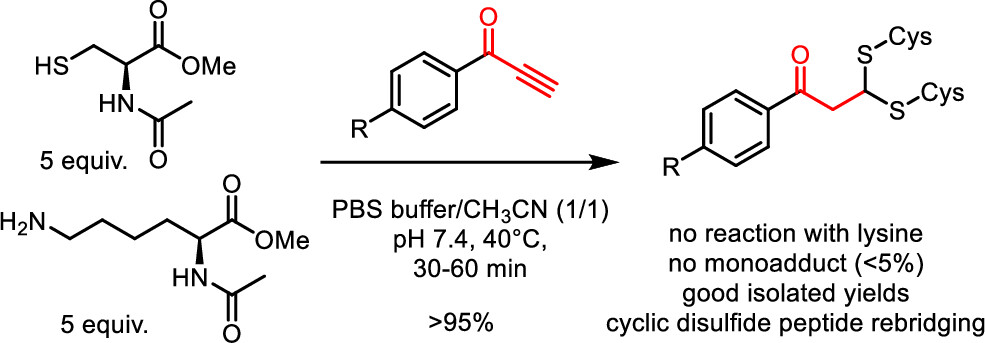
Simple 1-phenylpropynones undergo a selective double thia-Michael addition with thiols in buffered media, yielding an interesting dithioacetal linkage joining two thiols. The reactivity of various Michael-alkyne reagents is compared in this chemoselective, atom economical, and non-oxidative cross-linking of two thiols. The stability and chemical reactivity of the dithioacetal links are studied, and the utility of the disulfide targeting bioconjugation methodology is shown by the selective rebridging of native cyclic peptides after the reductive cleavage of their disulfide bridge.
Organic Letters, 26 (4), p895-899, 2024
https://doi.org/10.1021/acs.orglett.3c04160
71. Vinylogous Urea-Urethane Vitrimers: Accelerating and Inhibiting Network Dynamics through Hydrogen Bonding
Engelen S., Dolinski N. D., Chen C., Ghimire E., Lindberg C. A., Crolais A. E., Nitta N., Winne J. M., Rowan S. J., Du Prez F. E.
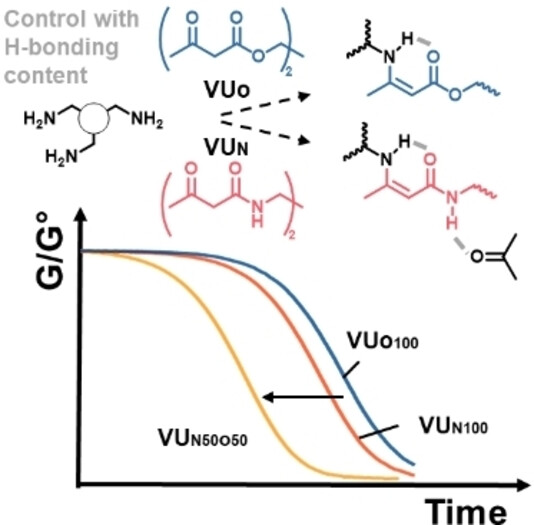
Vinylogous urethane (VUO) based polymer networks are widely used as catalyst-free vitrimers that show rapid covalent bond exchange at elevated temperatures. In solution, vinylogous ureas (VUN) undergo much faster bond exchange than VUO and are highly dynamic at room temperature. However, this difference in reactivity is not observed in their respective dynamic polymer networks, as VUO and VUN vitrimers prepared herein with very similar macromolecular architectures show comparable stress relaxation and creep behavior. However, by using mixtures of VUO and VUN linkages within the same network, the dynamic reactions can be accelerated by an order of magnitude. The results can be rationalized by the effect of intermolecular hydrogen bonding, which is absent in VUO vitrimers, but is very pronounced for vinylogous urea moieties. At low concentrations of VUN, these hydrogen bonds act as catalysts for covalent bond exchange, while at high concentration, they provide a pervasive vinylogous urea - urethane (VU) network of strong non-covalent interactions, giving rise to phase separation and inhibiting polymer chain dynamics. This offers a straightforward design principle for dynamic polymer materials, showing at the same time the possible additive and synergistic effects of supramolecular and dynamic covalent polymer networks.
Angewandte Chemie International Edition, 63 (9), pe20231841, 2024
https://doi.org/10.1002/anie.202318412
2023
70. Trialkylsulfonium-Based Reprocessable Polyurethane Thermosets
Scholiers V., Hendriks B., Maes S., Debsharma T., Winne J. M., Du Prez F. E.
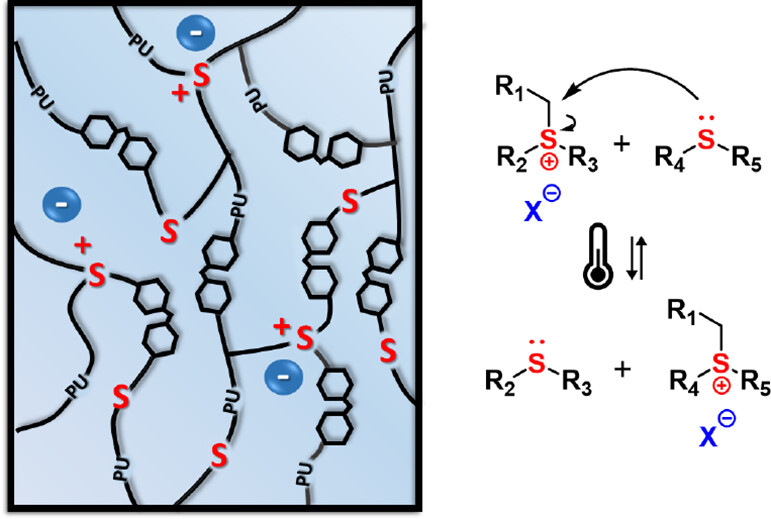
We report the development of a solvent-free protocol to produce colorless, highly transparent, and glassy polyurethane-based networks containing thioether bonds using commercially available building blocks. These polyurethane networks are converted into reprocessable networks by partial alkylation of the thioether bonds, giving dynamic trialkylsulfonium bonds that are able to exchange via transalkylation at elevated temperatures, thus inducing viscoelastic flow. Reprocessability of the trialkylsulfonium networks was demonstrated for three cycles without significant degradation of material properties. Interestingly, these materials were found to be highly processable at elevated temperatures (~140 °C) and showed excellent creep suppression up to 100 °C, a combination that is rare among dynamic covalent polymer networks. The suppression of creep can be further controlled by changing the alkylating additive. In addition, as a result of their excellent transparency and high clarity, we investigated their optical properties to assess their potential use in smart coatings and optical devices.
Macromolecules, 56 (23), p9559-9569, 2023
https://doi.org/10.1021/acs.macromol.3c01270
69. Dithioallyl Cations in Stereoselective Dearomative (3 + 2) Cycloadditions of Benzofurans: Mechanism and Synthetic Applications
Ryckaert B., Hullaert J., Van Hecke K. and Winne J. M.
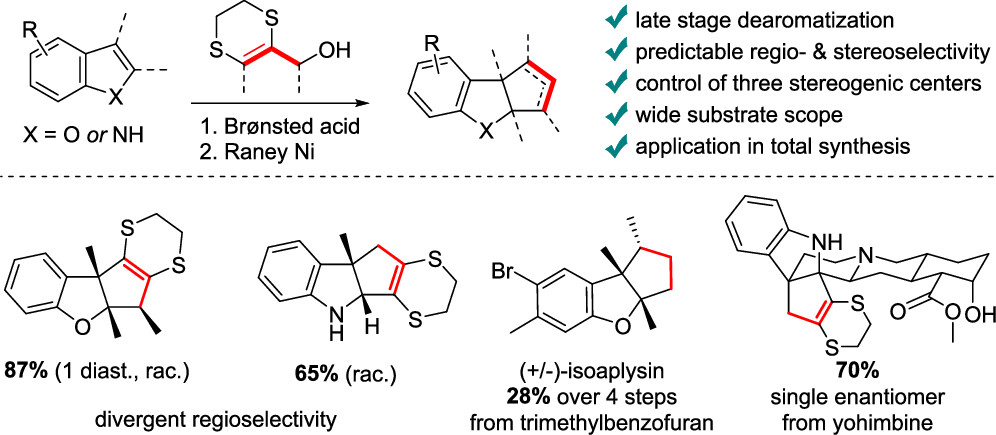
A stereoselective dearomative cyclopentannulation of benzofurans is reported. A previously reported dearomative (3 + 2) cycloaddition of indoles with 1,4-dithiane-fused allyl cations was found to lack stereoselectivity when more substituted cyclopentene rings are targeted. However, for benzofuran substrates, excellent levels of stereoselectivity were observed for the same allyl cation reagents under very similar reaction conditions. In this full account, we provide a mechanistic rationale and some design principles that govern the stereoselectivity of the intriguing dearomative transformations using dithioallyl cations and demonstrate how the stereoselectivity depends on electronic factors of the starting materials. The stereoselective methodology is also applied in a straightforward dearomative synthesis of the tricyclic sesquiterpenoid natural product aplysin and its analogues, starting from a simple benzofuran.
The Journal of Organic Chemistry, 20 (88), p14504-14514, 2023
https://doi.org/10.1021/acs.joc.3c01546
68. Dithioallyl cation (3 + 2) cycloadditions under aprotic reaction conditions: rapid access to spiro-fused cyclopentane scaffolds
Degroote F., Denoo B., Ryckaert B., Callebaut B., Van Hecke K., Hullaert J. and Winne J. M.
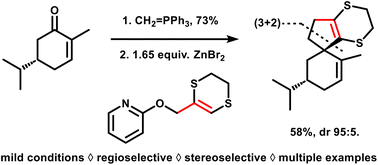
We report a general method to effect all-carbon (3 + 2) cycloadditions that can elaborate cyclopentenes from a range of olefins. The required dithioallyl cation reagents can be generated in a newly developed mild protocol starting from 2-allyloxypyridine precursors, thus avoiding the use of strong Brønsted acids. The novel method significantly expands the substrate scope, which now also includes acid-sensitive olefins, and thus enables the preparation of previously inaccessible spiro-fused scaffold types from simple and readily available starting materials.
Organic & Biomolecular Chemistry, 21 (40), p8117-8124, 2023
https://doi.org/10.1039/D3OB01273E
67. Site selective Gold(I)-Catalysed Benzylic C-H Amination via an Intermolecular Hydride Transfer to Triazolinediones
Bevernaege K., V Tzouras N., Poater A., Cavallo L., P Nolan S., Nahra F. and Winne J. M.

Triazolinediones are known as highly reactive dienophiles that can also act as electrophilic amination reagents towards enolisable C-H bonds (ionic pathway) or weak C-H bonds (free radical pathway). Here, we report that this C-H amination reactivity can be significantly extended and enhanced via gold(I)-catalysis. Under mild conditions, several alkyl-substituted aryls successfully undergo benzylic C-H aminations at room temperature. The remarkable site selectivity that is observed points towards strong electronic activation and deactivation effects, that go beyond a simple weakening of the C-H bond. The observed catalytic C-H aminations do not follow the expected trends for a free radical-type C-H amination and show complementarity to existing methods. Density functional theory (DFT) calculations and distinct experimental trends provide a clear mechanistic rationale for observed selectivity patterns, postulating a novel pathway for tria-zolinedione-induced aminations via a carbon-to-nitrogen hydride transfer.
Chemical Science, 14 (36), p9787-9794, 2023
https://pubs.rsc.org/en/content/articlelanding/2023/SC/D3SC03683A
66. Enhanced Viscosity Control in Thermosets Derived from Epoxy and Acrylate Monomers Based on Thermoreversible Aza-Michael Chemistry
Engelen S., Van Lijsebetten F., Aksakal R., Winne J. M., Du Prez F. E.
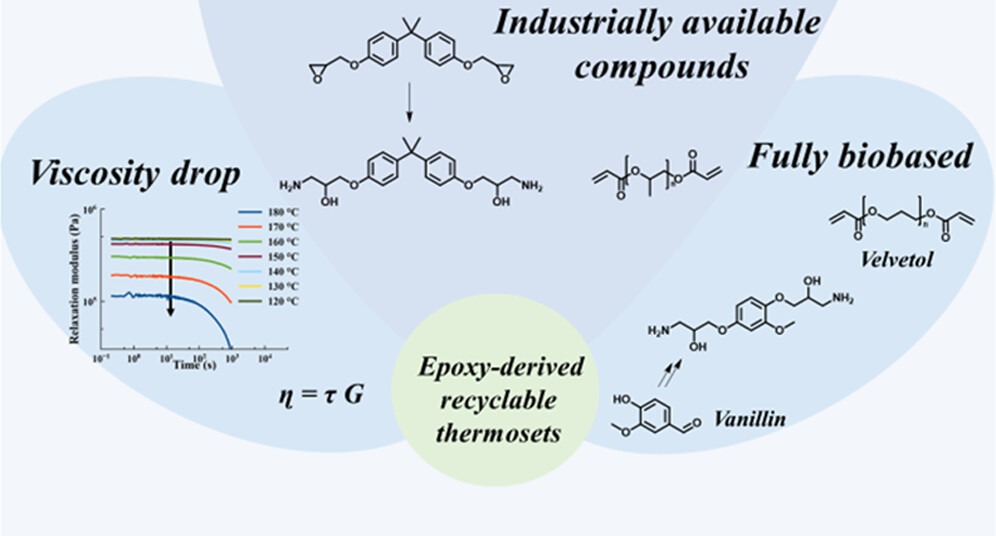
Epoxies, epoxy acrylates, and acrylic resins account for a large portion of the thermoset market, yet their inherently highly cross-linked nature prevents them from being reprocessed or recycled. Herein, we present a simple reversible epoxy-based curing strategy that can address these issues. First, an epoxy monomer is ring-opened with ammonia to result in β-hydroxyamines through a straightforward and scalable synthesis that we have demonstrated on multiple examples. The epoxy-derived amines are then cured with bisacrylates, creating dynamic covalent β-amino ester cross-linkages through a thermoreversible aza-Michael reaction. The resulting networks show a very pronounced drop in viscosity in the temperature region of 150-180 °C, which we were able to attribute mainly to significant de-cross-linking of the amine and acrylate moieties, rather than to activation of dynamic ester bond exchanges. Nevertheless, the materials do not fully liquify and retain their structural integrity as a result of the very fast amine-acrylate rebonding kinetics. As a result, cross-linked epoxy-based materials could be obtained with simultaneously enhanced (re)processability at high temperatures and strongly inhibited deformation at lower temperatures (<120 °C). The protocol is demonstrated for both petrochemically based building blocks (such as bisphenol A), as well as for fully biobased compounds (vanillin-epoxy and a Velvetol-based bisacrylate), showing its versatility. As a result of the widespread use of epoxy resins, the ease of implementation, and its interesting temperature window for debonding/rebonding, industrial applications can be foreseen for this thermoreversible curing strategy.
Macromolecules, 56 (17), p7055-7064, 2023
https://doi.org/10.1021/acs.macromol.3c01198
65. Plasmodesmata mediate cell-to-cell transport of brassinosteroid hormones
Wang Y., Perez-Sancho J., Platre M. P., Callebaut B., Smokvarska M., Ferrer K., Luo Y., Nolan T. M., Sato T., Busch W., Benfey P. N., Kvasnica M., Winne J. M., Bayer E. M., Vukašinovic N., Russinova E.
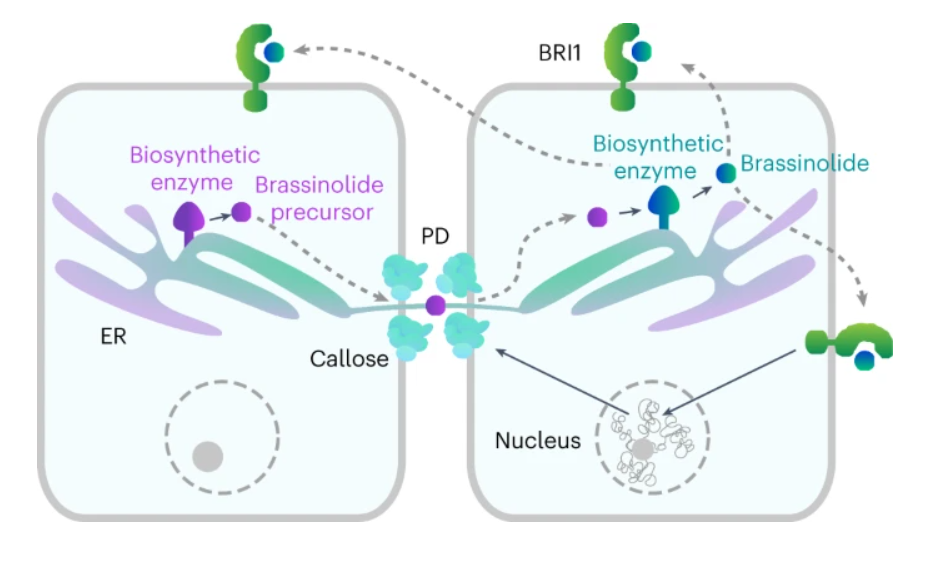
Brassinosteroids (BRs) are steroidal phytohormones that are essential for plant growth, development and adaptation to environmental stresses. BRs act in a dose-dependent manner and do not travel over long distances; hence, BR homeostasis maintenance is critical for their function. Biosynthesis of bioactive BRs relies on the cell-to-cell movement of hormone precursors. However, the mechanism of the short-distance BR transport is unknown, and its contribution to the control of endogenous BR levels remains unexplored. Here we demonstrate that plasmodesmata (PD) mediate the passage of BRs between neighboring cells. Intracellular BR content, in turn, is capable of modulating PD permeability to optimize its own mobility, thereby manipulating BR biosynthesis and signaling. Our work uncovers a thus far unknown mode of steroid transport in eukaryotes and exposes an additional layer of BR homeostasis regulation in plants.
Nature Chemical Biology, 19, p1331-1341, 2023
https://doi.org/10.1038/s41589-023-01346-x
64. Epoxy Adhesives with Reversible Hardeners: Controllable Thermal Debonding in Bulk and at Interfaces
Van Lijsebetten F., Maiheu T., Winne J. M., Du Prez F. E.
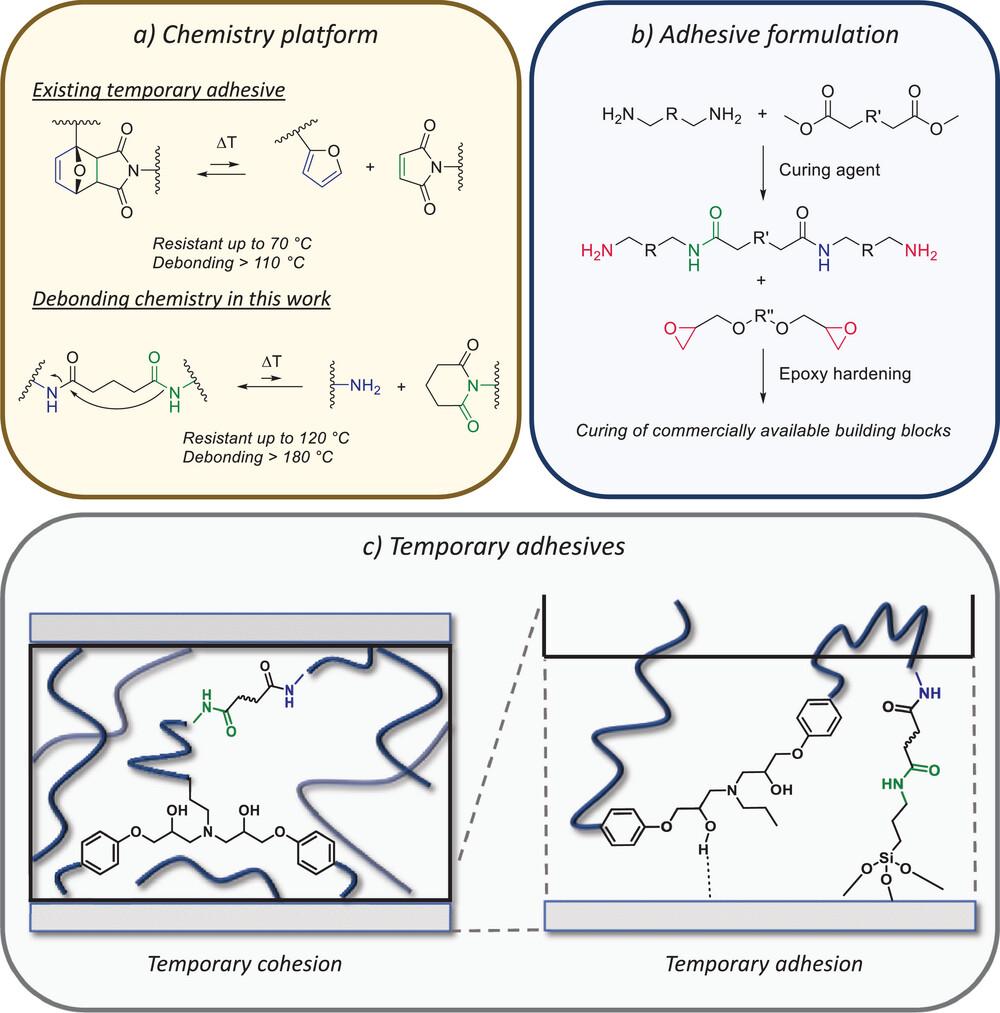
On-demand adhesive dismantling has the potential to improve multimaterial product recycling, but its implementation has been hampered by a critical trade-off between strong bonding and easy debonding. As a result, the temperature range in which these temporary adhesives can be used is relatively limited. Here, a new class of dynamic epoxy resins is reported that significantly extends this upper temperature limit and still achieves fast debonding. Specifically, two types of dynamic polyamidoamine curing agents for epoxy hardening are developed, being polysuccinamides (PSA) and polyglutaramides (PGA). As the dynamic debonding/rebonding process of PSA and especially PGA linkages is more thermally demanding and at the same time more thermally robust than previously reported dynamic covalent systems, the resulting materials can be triggered at high temperatures, and at the same time remain bonded over a wide temperature range. The versatility of the PSA and PGA dynamic adhesive curing system is demonstrated in classical bulk adhesive formulations, as well as in dynamic covalent linking to a PSA- or PGA-functionalized surface. As a result, an attractive drop-in strategy is achieved for producing debondable and rebondable epoxy adhesives, with high complementarity to existing adhesive resin technologies and applicable in an industrially relevant temperature window.
Advanced Materials, 35 (31), p2300802, 2023
https://doi.org/10.1002/adma.202300802
63. N-Sulfonyl Urethanes to Design Polyurethane Networks with Temperature-Controlled Dynamicity
Maes S., Van Lijsebetten F., Winne J. M. and Du Prez F. E.
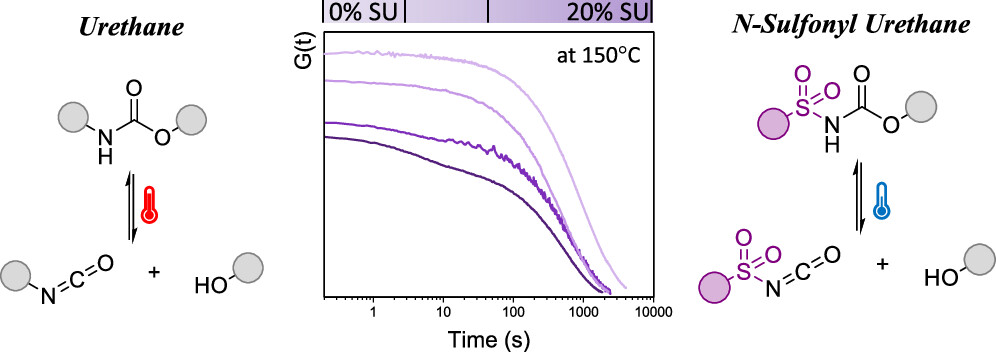
(Re)processing of cross-linked polyurethanes (PUs) is often energy intensive and inefficient since dissociation of urethane linkages at elevated temperatures generates highly reactive isocyanate moieties that can react with a wide range of nucleophiles. In this study, we first show with a small molecule study that the introduction of N-sulfonyl urethane bonds leads to dynamic covalent exchange reactions under much milder conditions compared to regular urethane groups. Then, these exchangeable N-sulfonyl urethane motifs have been introduced, in relatively small amounts (5, 10, and 20%), in a cross-linked PU matrix in an attempt to facilitate plastic flow at lower temperatures. Rheological analysis of the elastomeric dissociative networks revealed an interesting double relaxation behavior, even for temperatures between 150 and 100 °C, which could be described by a Maxwell model with two elements, which can be related to the activated and less activated urethane bonds. Finally, the (re)processability of these sulfonyl urethanes containing PUs was demonstrated through multiple cutting and hot pressing cycles and the corresponding materials showed a good retention of thermal properties.
Macromolecules, 56, p1934-1944, 2023
https://pubs.acs.org/doi/full/10.1021/acs.macromol.2c02456
62. 1,4-Dithianes: attractive C2-building blocks for the synthesis of complex molecular architectures
Ryckaert B., Demeyere E., Degroote F., Janssens H. and Winne J. M.

This review covers the synthetic applications of 1,4-dithianes, as well as derivatives thereof at various oxidation states. The selected examples show how the specific heterocyclic reactivity can be harnessed for the controlled synthesis of carbon–carbon bonds. The reactivity is compared to and put into context with more common synthetic building blocks, such as 1,3-dithianes and (hetero)aromatic building blocks. 1,4-Dithianes have as yet not been investigated to the same extent as their well-known 1,3-dithiane counterparts, but they do offer attractive transformations that can find good use in the assembly of a wide array of complex molecular architectures, ranging from lipids and carbohydrates to various carbocyclic scaffolds. This versatility arises from the possibility to chemoselectively cleave or reduce the sulfur-heterocycle to reveal a versatile C2-synthon.
Beilstein Journal of Organic Chemistry 19:12, 19, p115-132, 2023
https://www.beilstein-journals.org/bjoc/articles/19/12
2022
61. A Highly Dynamic Covalent Polymer Network without Creep: Mission Impossible?
Van Lijsebetten F., Debsharma T., Winne J. M. and Du Prez F. E.
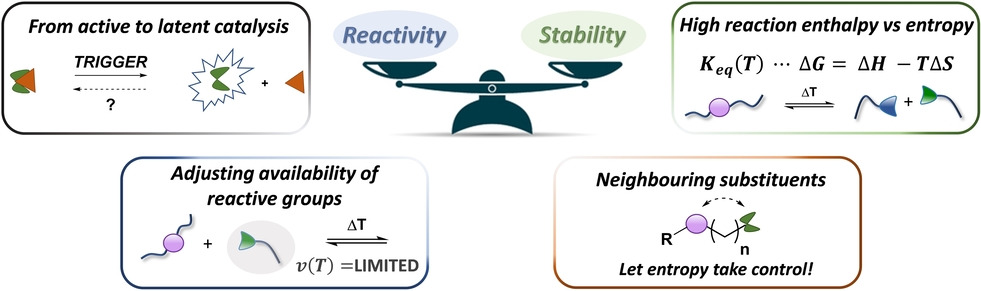
Dynamic covalent polymer networks provide an interesting solution to the challenging recyclability of thermosets and elastomers. One of the remaining design constraints, however, is balancing thermal reprocessability in the form of material flow with dimensional stability during use. As a result, many chemistries are being investigated in order to improve bond reactivity control and material robustness. This Minireview highlights a number of promising concepts, with a particular emphasis on disconnecting chemical reactivity in low and high temperature regimes to obtain creep resistant, yet highly dynamic polymer networks. In addition, we will highlight the impact of sharp reactivity changes when applying extrapolation-based approaches during rheological analysis. As a result, we are confident that abandoning the myth of permanent reactivity will aid in the development of sustainable polymeric materials that can truly combine the benefits of thermoplastic and thermoset behaviour.
Angewandte Chemie International Edition, 61, pe202210405, 2022
https://onlinelibrary.wiley.com/doi/full/10.1002/anie.202210405
60. Characterising different molecular landscapes in dynamic covalent networks
Van Lijsebetten F., De Bruycker K., Van Ruymbeke E., Winne J. M. and Du Prez F. E.
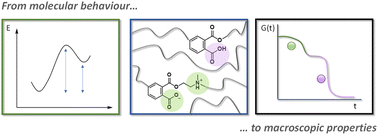
Dynamic covalent networks present a unique opportunity to exert molecular-level control on macroscopic material properties, by linking their thermal behaviour to the thermodynamics and kinetics of the underlying chemistry. Yet, existing methods do not allow for the extraction and analysis of the influence of local differences in chemical reactivity caused by available reactants, catalysts, or additives. In this context, we present a rheological paradigm that allows us to correlate the composition of a reactive polymer segment to a faster or slower rate of network rearrangement. We discovered that a generalised Maxwell model could separate and quantify the dynamic behaviour of each type of reactive segment individually, which was crucial to fully comprehend the mechanics of the final material. More specifically, Eyring and Van 't Hoff analysis were used to relate possible bond catalysis and dissociation to structural changes by combining statistical modelling with rheology measurements. As a result, precise viscosity changes could be measured, allowing for accurate comparison of various dynamic covalent network materials, including vitrimers and dissociative networks. The herein reported method therefore facilitated the successful analysis of virtually any type of rate-enhancing effect and will allow for the design of functional and fast (re)processable materials, as well as improve our ability to predict and engineer their properties for future applications.
Chemical Science, 13, p12865-12875, 2022
https://pubs.rsc.org/en/content/articlehtml/2022/sc/d2sc05528g
59. Masked Primary Amines for a Controlled Plastic Flow of Vitrimers
Van Lijsebetten F., De Bruycker K., Winne J. M. and Du Prez F. E.
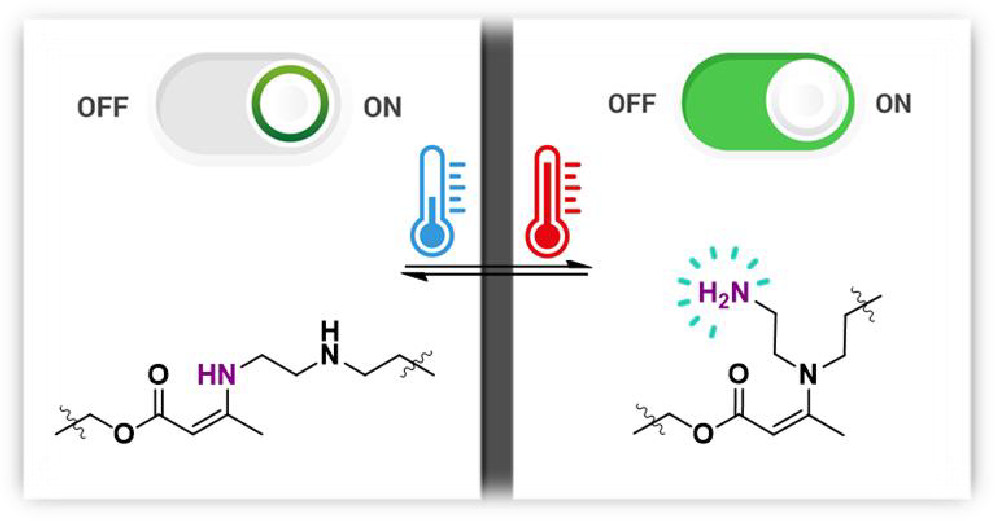
We present a simple method for increasing the reprocessability of vinylogous urethane (VU) vitrimers while decreasing the possibility of creep deformation at lower temperatures. In particular, varying amounts of triethylenetetramine were added as a comonomer to the curing VU formulation to ensure that all of the primary amines reacted to form enaminone cross-links, resulting in a network without reactive primary amine chain-ends. As a result, transamination was significantly slowed down because secondary amines are much less reactive to VU exchange. On the other hand, at higher temperatures, pendent primary amines can be released via a dynamic, endothermic exchange with a nearby less-reactive secondary amine, thereby (re)activating material flow. As a result, ambivalent viscoelastic behavior could be achieved without depolymerization by dynamically releasing pendent primary amines from vinylogous urethane polymer chains. Through careful comonomer selection, VU vitrimers with low viscosity at processing temperatures and at the same time high viscosity at service temperatures could be prepared without the use of catalysts or additives, leveraging the synergistic effects of mildly reactive functionalities through neighboring group participation.
ACS Macro Letters, 11, p919-924, 2022
https://pubs.acs.org/doi/full/10.1021/acsmacrolett.2c00255
58. Dearomative (3 + 2) Cycloadditions of Unprotected Indoles
Ryckaert B., Hullaert J., Van Hecke K., Winne J. M.
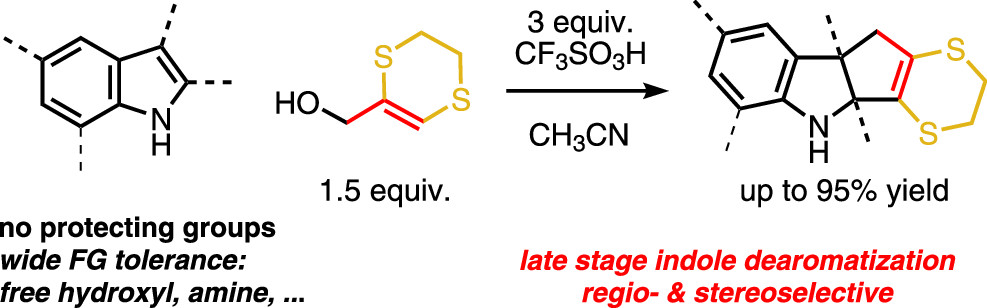
The (3 + 2) cycloaddition of various indoles with a dithioallyl cation affords dearomatized cyclopentannulated adducts, with complete control of regioselectivity and excellent chemo- and diastereoselectivity. The success of the reaction critically relies on the use of an excess of very strong Brønsted acid, which paradoxically prevents carbocationic side reactions. The reaction tolerates sensitive functionalities such as basic amines or free hydroxyls, and we demonstrate its use in late stage derivatization of highly functionalized, unprotected indoles.
Organic Letters, 24 (23), p4119-4123, 2022
https://pubs.acs.org/doi/full/10.1021/acs.orglett.2c01214
57. Triazolinedione protein modification: from an overlooked off-target effect to a tryptophan-based bioconjugation strategy
Decoene K. W., Unal K., Staes A., Zwaenepoel O., Gettemans J., Gevaert K., Winne J. M. and Madder A.
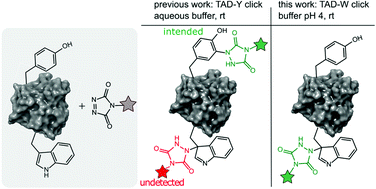
Labelling of tyrosine residues in peptides and proteins has been reported to selectively occur via a 'tyrosine-click reaction with triazolinedione reagents (TAD). However, we here demonstrate that TAD reagents are actually not selective for tyrosine and that tryptophan residues are in fact also labelled with these reagents. This off-target labelling remained under the radar as it is challenging to detect these physiologically stable but thermally labile modifications with the commonly used HCD and CID MS/MS techniques. We show that selectivity of tryptophan over tyrosine can be achieved by lowering the pH of the aqueous buffer to effect selective Trp-labelling. Given the low relative abundance of tryptophan compared to tyrosine in natural proteins, this results in a new site-selective bioconjugation method that does not rely on enzymes nor unnatural amino acids and is demonstrated for peptides and recombinant proteins.
Chemical Science, 13, p5390-5397, 2022
https://pubs.rsc.org/en/content/articlehtml/2022/sc/d1sc06942j
56. Synthesis of Cyclopropyl Pinacol Boronic Esters from Dibromocyclopropanes
Neouchy Z., Hullaert J., Verhoeven J., Meerpoel L., Thuring J., Verniest G., Winne J. M.
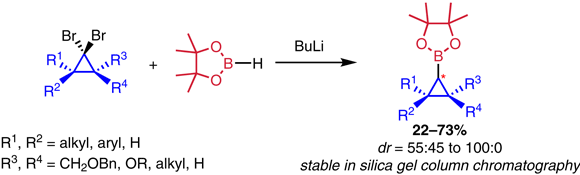
The synthesis of cyclopropyl pinacol boronic esters from dibromocyclopropanes via Matteson-Pasto rearrangement is reported. The method is readily scalable and shows limited levels of stereoinduction, with a selectivity that is in part complementary to that observed in existing stereoselective borylcyclopropanation strategies. The method can be used to rapidly access borylcyclopropanes as interesting building blocks for diversely functionalized cyclopropanes.
Synlett, 33 (08), p759-766, 2022
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0037-1610794
55. Suppressing Creep and Promoting Fast Reprocessing of Vitrimers with Reversibly Trapped Amines
Van Lijsebetten F., De Bruycker K., Spiesschaert Y., Winne J. M., Du Prez F. E.
Viscous deformation in elastomeric vitrimers is suppressed by temporarily trapping a reactive side chain in the polymer backbone. Upon heating, the network defect is recovered allowing for fast exchange. This offers a versatile strategy to simultaneously achieve fast reprocessing, while maintaining the strong dimensional stability of vitrimers.
Angewandte Chemie International Edition, 61 (9), p1433-7851, 2022
https://onlinelibrary.wiley.com/doi/10.1002/anie.202113872
2021
54. Stereodivergent Synthesis of Biologically Active Spironucleoside Scaffolds via Catalytic Cyclopropanation of 4-exo-Methylene Furanosides
Neouchy Z., Verhoeven J., Kong H., Zhao Y., Wang w., Brambilla M., Van Hecke K., Meerpoel L., Thuring J., Verniest G., Winne J. M.
Cyclopropane fusion of the only rotatable carbon-carbon bond in furanosyl nucleosides (i.e., exocyclic 4'-5') is a powerful design strategy to arrive at conformationally constrained analogues. Herein, we report a direct stereodivergent route toward the synthesis of the four possible configurations of 4-spirocyclopropane furanoses, which have been transformed into the corresponding 4'-spirocyclic adenosine analogues. The latter showed differential inhibition of the protein methyltransferase PRMT5-MEP50 complex, with one analogue inhibiting more effectively than adenosine itself, demonstrating the utility of rationally probing 4'-5' side chain orientations.
The Journal of Organic Chemistry, 86 (23), p17344-17361, 2021
https://pubs.acs.org/doi/10.1021/acs.joc.1c01611
53. Regio- and Stereoselective Synthesis of C-4' Spirocyclobutyl Ribofuranose Scaffolds and Their Use as Biologically Active Nucleoside Analogues
Jouffroy L., Verhoeven J., Brambilla M., Verniest G., Kong H., Zhao Y., Wang W., Meerpoel L., Thuring J., Winne J. M.
Novel C-4',C-5' cyclobutane-fused spirocyclic ribonucleoside analogues were prepared. Thermal [2 + 2] cycloaddition between dichloroketene and readily derived 4'-exo-methylene furanoses afforded a first entry to the required constrained ribofuranoses, relying on a carbonyl transposition sequence. Alternatively, an unusual stereoselective ionic [2 + 2] cycloaddition using methyl propiolate promoted by methylaluminoxane gave a complementary, more direct approach to such ribofuranoses. Further conversion to the constrained adenosine analogues revealed promising structure-dependent inhibition of the protein methyltransferase PRMT5:MEP50 complex in the (sub)micromolar range.
Organic Letters, 23 (22), p8828-8833, 2021
https://pubs.acs.org/doi/full/10.1021/acs.orglett.1c03334
52. Polyaddition Synthesis Using Alkyne Esters for the Design of Vinylogous Urethane Vitrimers
Spiesschaert Y., Danneels J., Herck N. V., Guerre M., Acke G., Winne J. and Prez F. D.
Vitrimers are a subclass of covalent adaptable networks which introduce reshapeability and recyclability in thermoset materials while maintaining a high degree of chemical resistance and dimensiona...
Macromolecules, 54 (17), p7931-7942, 2021
https://pubs.acs.org/doi/abs/10.1021/acs.macromol.1c01049
51. Reprocessing of Covalent Adaptable Polyamide Networks through Internal Catalysis and Ring-Size Effects
Lijsebetten F. V., Spiesschaert Y., Winne J. M. and Prez F. E. D.
Here, we report the introduction of internally catalyzed amide bonds to obtain covalent adaptable polyamide networks that rely on the dissociation equilibrium between dicarboxamides and imides. Whi...
Journal of the American Chemical Society, , pjacs.1c07360, 2021
https://pubs.acs.org/doi/abs/10.1021/jacs.1c07360
2020
50. Stereoselective Gold(I)-Catalyzed Vinylcyclopropanation via Generation of a Sulfur-Substituted Vinyl Carbene Equivalent
Yuan T., Ryckaert B., Hullaert J., Van Hecke K., and Winne J. M.
A stereoselective gold(I)‐catalyzed vinylcyclopropanation of alkenes has been developed. A gold-coordinated cationic vinyl carbene species, readily generated via a rearrangement of the ethylenedithioacetal of propargyl aldehyde, reacts with a wide range of alkenes to afford thio-substituted vinylcyclopropanes. The gold-catalyzed vinyl cyclopropanation proceeds under mild conditions at room temperature and is generally selective for the formation of cis‐substituted cyclopropanes. The reaction allows the formal introduction of a 'naked' vinyl carbene, by subsequent chemoselective hydrodesulfurisation of the ethylenedithio-bridge. The synthetic utility of the new method is demonstrated by a short, racemic formal synthesis of the alkaloid cephalotaxin, hinging on a key vinyl cyclopropane‐cyclopentene rearrangement.
Angewandte Chemie International Edition, 60 (8), p4070-4074, 2020
https://onlinelibrary.wiley.com/doi/10.1002/anie.202012664
49. Influence of the polymer matrix on the viscoelastic behaviour of vitrimers
Spiesschaert Y., Taplan C., Stricker L., Guerre M., Winne J. M. and Du Prez F. E.
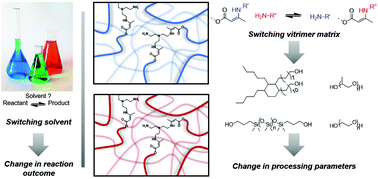
Vitrimer materials are a rapidly growing field of research, in which still many fundamental aspects of material design remain to be explored. In this study, we report a systematic study about the effect of the choice of the matrix on a dynamic covalent bond exchange reaction in a polymer network. In some way, this investigation follows the logic of a 'solvent effect' study that is typical for the development of an organic reaction or synthetic method in solution. Thus, this work constitutes a study of matrix effects on the viscoelastic properties of vitrimers, in particular with regard to their stress-relaxation behaviour. For that purpose, the dynamic transamination within vinylogous urethanes (VU) vitrimers, derived from a wide range of different commercially available diols, was chosen as vitrimer chemistry reaction platform. Additionally, the influence of the molecular weight, and thus the cross-link density, was also investigated using two oligomeric diols of different molecular weights. It was found that changing the molecular weight resulted in vitrimers with significantly different activation energies (from 68 to 149 kJ mol-1) and relaxation times (from 7 to 230 min at 140 °C). These remarkable results suggest that both the molecular weight and the nature of the polymer matrix have a large influence on the resulting viscoelastic properties. However, no straightforward prediction of relaxation times or activation energies is possible at this stage, and it becomes clear that many more structure-reactivity studies will be required to arrive at a more general understanding of the factors that underly vitrimer properties. This journal is
Polymer Chemistry, 11 (33), p5377-5385, 2020
https://pubs.rsc.org/en/content/articlehtml/2020/py/d0py00114g
48. Double neighbouring group participation for ultrafast exchange in phthalate monoester networks
Delahaye M., Tanini F., Holloway J. O., Winne J. M. and Du Prez F. E.
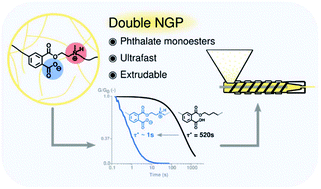
Phthalate monoesters (PMEs) were recently introduced as a simple dynamic covalent bond for implementation in covalent adaptable networks (CANs), which undergo rapid transesterifications in the absence of catalysts, due to the neighbouring group participation (NGP) of a carboxylic acid moiety. In this work, it is shown that the PME transesterification can be very significantly accelerated by the presence of another neighbouring group on the reactive alcohol moieties. The kinetic effects are demonstrated using a short model study of PMEs with different substituents at the β-carbon position, showing a remarkable acceleration for alcohols containing tertiary amines on the β-carbon. Following the model study, materials were synthesised by a (partial) replacement of the conventionally used diol with a β-amino-diol, leading to the formation of networks with an increased Tg and Young's-modulus, which is rationalised as a result of the formation of an ionic network (COO- and NHR3+). Stress relaxation experiments show a decrease in relaxation times by a factor of 500, compared to similar networks derived from non-amine-substituted hydroxyl monomers. This ultrafast relaxation, enabled by a double NGP, resulted in CANs that show potential to be processed through extrusion while maintaining their overall network connectivity.
Polymer Chemistry, 11 (32), p5207-5215, 2020
https://pubs.rsc.org/en/content/articlehtml/2020/py/d0py00681e
47. Vitrimers: directing chemical reactivity to control material properties
Guerre M., Taplan C., Winne J. M. and Du Prez F. E.
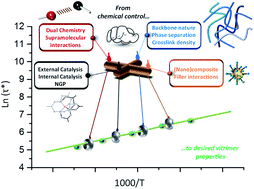
The development of more sustainable materials with a prolonged useful lifetime is a key requirement for a transition towards a more circular economy. However, polymer materials that are long-lasting and highly durable also tend to have a limited application potential for re-use. This is because such materials derive their durable properties from a high degree of chemical connectivity, resulting in rigid meshes or networks of polymer chains with a high intrinsic resistance to deformation. Once such polymers are fully synthesised, thermal (re)processing becomes hard (or impossible) to achieve without damaging the degree of chemical connectivity, and most recycling options quickly lead to a drop or even loss of material properties. In this context, both academic and industrial researchers have taken a keen interest in materials design that combines high degrees of chemical connectivity with an improved thermal (re)processability, mediated through a dynamic exchange reaction of covalent bonds. In particular vitrimer materials offer a promising concept because they completely maintain their degree of chemical connectivity at all times, yet can show a clear thermally driven plasticity and liquid behavior, enabled through rapid bond rearrangement reactions within the network. In the past decade, many suitable dynamic covalent chemistries were developed to create vitrimer materials, and are now applicable to a wide range of polymer matrices. The material properties of vitrimers, however, do not solely rely on the chemical structure of the polymer matrix, but also on the chemical reactivity of the dynamic bonds. Thus, chemical reactivity considerations become an integral part of material design, which has to take into account for example catalytic and cross-reactivity effects. This mini-review will aim to provide an overview of recent efforts aimed at understanding and controlling dynamic cross-linking reactions within vitrimers, and how directing this chemical reactivity can be used as a handle to steer material properties. Hence, it is shown how a focus on a fundamental chemical understanding can pave the way towards new sustainable materials and applications.
Polymer Chemistry, 11 (19), p4855-4870, 2020
https://pubs.rsc.org/en/content/articlelanding/2020/SC/D0SC01069C#!divAbstract
46. Dynamic Curing Agents for Amine-Hardened Epoxy Vitrimers with Short (Re)processing Times
Spiesschaert Y., Guerre M., De Baere I., Van Paepegem W., Winne J. M. and Du Prez F. E.
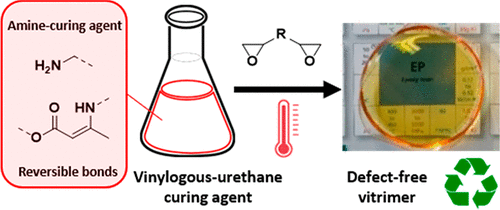
This work presents a straightforward strategy to introduce highly dynamic and adaptable cross-links into common epoxy resin formulations. For this, an oligomeric amine-based curing agent containing vinylogous urethane (VU) bonds was developed. This novel polyfunctional amine curing agent can be used as a drop-in solution for existing epoxy resin technologies, resulting in transparent, rigid, and, at the same time, highly reprocessable catalyst-free epoxy vitrimers. The oligomeric VU-curing agents are prepolymerized via a straightforward condensation reaction between acetoacetates extended with different classical amine monomers and epoxy hardeners. It is found that vitrimer properties can be readily introduced into these epoxy formulations by converting <50 mol % of the hardener's amine functionalities into dynamic vinylogous urethane bonds. In this way, epoxy vitrimers can be obtained with material properties comparable to ones of the VU-free epoxy formulations. In addition, remarkably short processing times are observed in the absence of any catalyst, and the material displayed very short stress relaxation times and good recyclability, actually representing the most performant VU-based vitrimers so far. Furthermore, a proof of concept for its use in obtaining glass-fiber-reinforced epoxy composites is also presented.
Macromolecules, 53 (7), p2485-2495, 2020
https://dx.doi.org/10.1021/acs.macromol.9b02526
45. Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol-yne Cross-Linking
Van Herck N., Maes D., Ünal K., Guerre M., Winne J. M., Du Prez F. E.
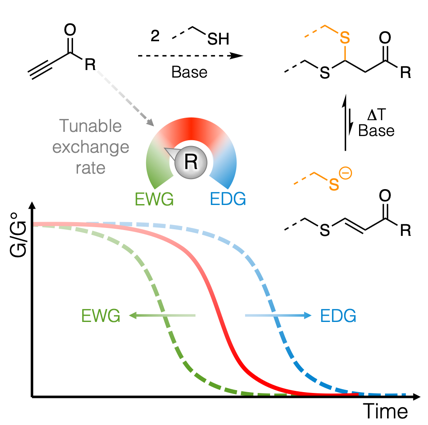
The design of covalent adaptable networks (CANs) relies on the ability to trigger the rearrangement of bonds within a polymer network. Simple activated alkynes are now used as versatile reversible cross-linkers for thiols. The click-like thiol-yne cross-linking reaction readily enables network synthesis from polythiols through a double Michael addition with a reversible and tunable second addition step. The resulting thioacetal cross-linking moieties are robust but dynamic linkages. A series of different activated alkynes have been synthesized and systematically probed for their ability to produce dynamic thioacetal linkages, both in kinetic studies of small molecule models, as well as in stress relaxation and creep measurements on thiol-yne-based CANs. The results are further rationalized by DFT calculations, showing that the bond exchange rates can be significantly influenced by the choice of the activated alkyne cross-linker.
Angewandte Chemie International Edition, 59 (9), p3609-3617, 2020
https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201912902
44. Fast processing of highly crosslinked, low-viscosity vitrimers
Taplan C., Guerre M., Winne J. M. and Du Prez F. E.
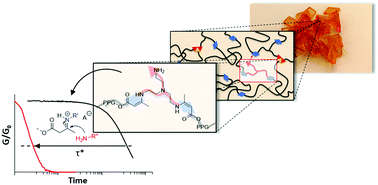
Here we describe a rational approach to go beyond the current processability limits of vitrimer materials, with a demonstration of low-viscosity fast processing of highly crosslinked permanent networks. Vitrimers are a recently introduced class of polymer networks with a unique glass-like viscoelastic behavior, in which bond exchange reactions govern the macroscopic material flow. The restricted chain mobility, only enabled by chemical exchanges, typically limits the use of continuous processing techniques, such as extrusion or injection moulding. Herein, we outline a straightforward materials design approach, taking into account both the effect of minor additives on the chemistry of bond rearrangement as well as the macromolecular architecture of the vitrimeric network. These combined effects are demonstrated to work in an additive fashion, culminating in stress relaxation times below 1 s at 150 °C. The observed rapid bond exchanges in permanent networks result in an unprecedented control of the polymer material behavior, where the material flow is still dominated by chemical exchanges, but only marginally limited by the chemical exchange rate, overcoming the challenges encountered so far in continuous processing of highly crosslinked vitrimeric systems.
Materials Horizons, 7 (1), p104-110, 2020
https://pubs.rsc.org/en/content/articlehtml/2020/mh/c9mh01062a
2019
43. Synthetic biomolecules: from blind watchmakers to synthetic biologists
Winne J. and Madder A.
Covalent adaptable networks (CANs) often make use of highly active external catalysts to provide swift exchange of the dynamic chemical bonds. Alternatively, milder species can act as internal catalysts when covalently attached to the matrix and in close proximity to the dynamic bonds. In this context, we introduce the dynamic exchange of phthalate monoesters as a novel chemistry platform for covalent adaptable networks. A low-molecular-weight (MW) model study shows that these monoesters undergo fast transesterification via a dissociative mechanism, caused by internal catalysis of the free carboxylic acid, which reversibly forms an activated phthalic anhydride intermediate. Using this dynamic chemistry, a wide series of CANs with a broad range of properties have been prepared by simply curing a mixture of diols and triols with bifunctional phthalic anhydrides. The dynamic nature of the networks was confirmed via recycling experiments for multiple cycles and via stress relaxation using rheology. The networks proved to be resistant to deformation but showed a marked temperature response in their rheological behavior, related to the swift exchange reactions that have a high activation energy (120 kJ/mol). While densely cross-linked and hydrolytically stable polyester networks with low soluble fractions can be obtained, we found that, by swelling the networks in a hot solvent, a gel-to-sol transition happened, which resulted in the full dissolution of the network.
Journal of the American Chemical Society, 52 (38), pA3-A5, 2019
https://www.sciencedirect.com/science/article/pii/S1367593119301115?via%3Dihub
42. Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry
Delahaye M., Winne J. M. and Du Prez F. E.
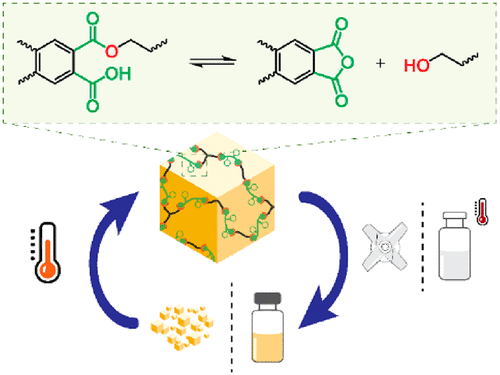
Covalent adaptable networks (CANs) often make use of highly active external catalysts to provide swift exchange of the dynamic chemical bonds. Alternatively, milder species can act as internal catalysts when covalently attached to the matrix and in close proximity to the dynamic bonds. In this context, we introduce the dynamic exchange of phthalate monoesters as a novel chemistry platform for covalent adaptable networks. A low-molecular-weight (MW) model study shows that these monoesters undergo fast transesterification via a dissociative mechanism, caused by internal catalysis of the free carboxylic acid, which reversibly forms an activated phthalic anhydride intermediate. Using this dynamic chemistry, a wide series of CANs with a broad range of properties have been prepared by simply curing a mixture of diols and triols with bifunctional phthalic anhydrides. The dynamic nature of the networks was confirmed via recycling experiments for multiple cycles and via stress relaxation using rheology. The networks proved to be resistant to deformation but showed a marked temperature response in their rheological behavior, related to the swift exchange reactions that have a high activation energy (120 kJ/mol). While densely cross-linked and hydrolytically stable polyester networks with low soluble fractions can be obtained, we found that, by swelling the networks in a hot solvent, a gel-to-sol transition happened, which resulted in the full dissolution of the network.
Journal of the American Chemical Society, 141 (38), p15277-15287, 2019
https://pubs.acs.org/doi/10.1021/jacs.9b07269
41. Disruption of endocytosis through chemical inhibition of clathrin heavy chain function
Dejonghe W., Sharma I., Denoo B., De Munck S., Lu Q., Mishev K., Bulut H., Mylle E., De Rycke R., Vasileva M., Savatin D. V., Nerinckx W., Staes A., Drozdzecki A., Audenaert D., Yperman K., Madder A., Friml J., Van Damme D., Gevaert K., Haucke V., Savvides S. N., Winne J. and Russinova E.
Clathrin-mediated endocytosis (CME) is a highly conserved and essential cellular process in eukaryotic cells, but its dynamic and vital nature makes it challenging to study using classical genetics tools. In contrast, although small molecules can acutely and reversibly perturb CME, the few chemical CME inhibitors that have been applied to plants are either ineffective or show undesirable side effects. Here, we identify the previously described endosidin9 (ES9) as an inhibitor of clathrin heavy chain (CHC) function in both Arabidopsis and human cells through affinity-based target isolation, in vitro binding studies and X-ray crystallography. Moreover, we present a chemically improved ES9 analog, ES9-17, which lacks the undesirable side effects of ES9 while retaining the ability to target CHC. ES9 and ES9-17 have expanded the chemical toolbox used to probe CHC function, and present chemical scaffolds for further design of more specific and potent CHC inhibitors across different systems.
Nature Chemical Biology, 15 (6), p641-649, 2019
https://www.nature.com/articles/s41589-019-0262-1
40. Filler reinforced polydimethylsiloxane-based vitrimers
Spiesschaert Y., Guerre M., Imbernon L., Winne J. M. and Du Prez F. E.
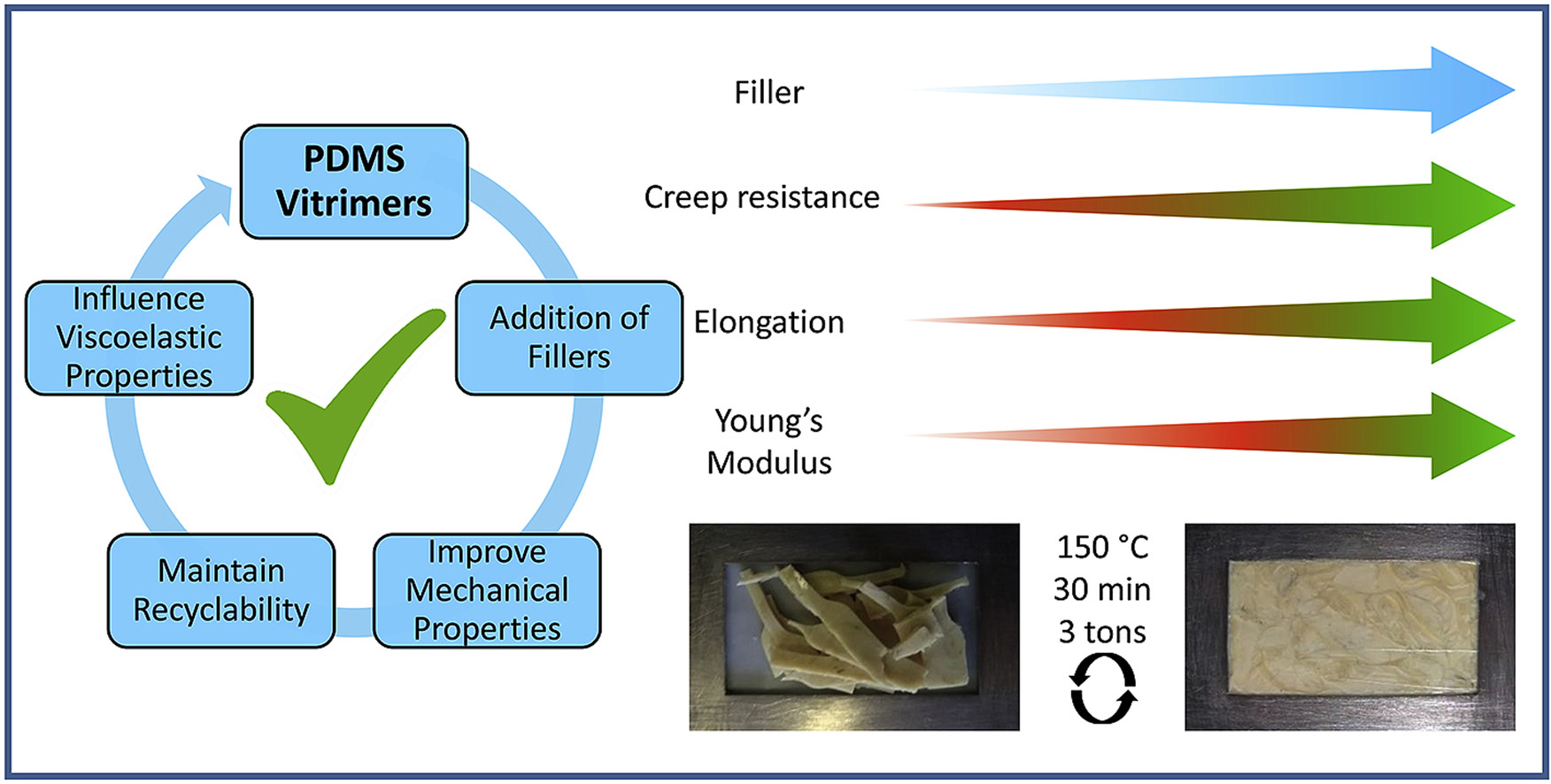
Vitrimers are a type of covalent adaptable networks that use an associative mechanism resulting in the ability to be thermally processed without losing their network integrity, even at elevated temperatures. Here we describe the synthesis of vinylogous urethane-based thermally stable, reinforced polydimethylsiloxane vitrimers with a special emphasis on the influence of the filler's acidic or basic functionalization on relaxation times. The vitrimer design is based on a straightforward condensation reaction between amino terminated polydimethylsiloxane and a trifunctional acetoacetate. The addition of fillers improves the mechanical properties, reduces the soluble fraction, preserves processability and, depending on the amount of filler added, maintains the ability to be recycled, without loss of properties. Furthermore, the relaxation times can be slightly tuned by using acidic or basic fillers, whilst keeping the corresponding material properties. These results show that fillers can be used as both reinforcing agents and catalysts in vinylogous urethane vitrimer systems.
Polymer, 172, p239-246, 2019
https://www.sciencedirect.com/science/article/pii/S0032386119303076?via%3Dihub
39. An Intramolecular Cycloaddition Approach to the Kauranoid Family of Diterpene Metabolites
Callebaut B., Hullaert J., Van Hecke K. and Winne J. M.
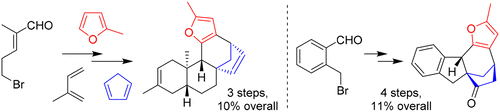
Synthetic studies toward the ent-kauranoid family of diterpene natural products are reported. An intramolecular (4 + 3) cycloaddition allows the direct elaboration of diverse natural product frameworks, encompassing a challenging bicyclo[3.2.1]octane core. The established routes comprise only a few synthetic operations (3–5 steps), transforming a range of simple starting materials into the tetracyclic scaffolds that are commonly found in many ent-kaurene metabolites.
Organic Letters, 21 (1), p310-314, 2019
http://pubs.acs.org/doi/10.1021/acs.orglett.8b03810
2018
38. The Elusive Seven-Membered Cyclic Imino Ether Tetrahydrooxazepine
Verbraeken B., Hullaert J., Van Guyse J., Van Hecke K., Winne J. and Hoogenboom R.
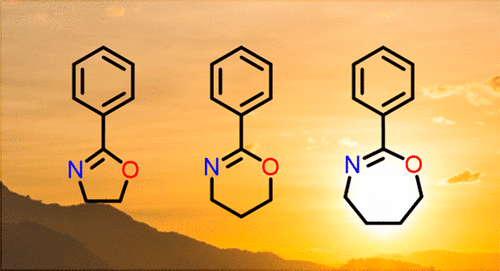
Cyclic imino ether heterocycles are used as ligands in transition metal catalysis, in various drugs and as reactive monomers in living cationic ring-opening polymerization (CROP). While five- and six-membered cyclic imino ethers, i.e. 2-oxazolines and 4,5-dihydro-1,3-oxazines, have extensively been studied in these areas, their seven-membered ring counterparts have remained unexplored. Herein, we report the synthesis of 2-phenyl-4,5,6,7-tetrahydro-1,3-oxazepine allowing reassignment of the earlier, incorrectly reported 4,5,6,7-tetrahydro-1,3-oxazepines as their N-acylated pyrrolidine isomers. Finally, we also report a comparison of the CROP reactivity of a homologous series of cyclic imino ethers with a 2-carbon, 3-carbon, and 4-carbon methylene bridge, revealing a remarkable ring size effect.
Journal of the American Chemical Society, 140 (50), p17404-17408, 2018
https://pubs.acs.org/doi/10.1021/jacs.8b10918
37. Fluorinated Vitrimer Elastomers with a Dual Temperature Response
Guerre M., Taplan C., Nicolaÿ R., Winne J. M. and Du Prez F. E.
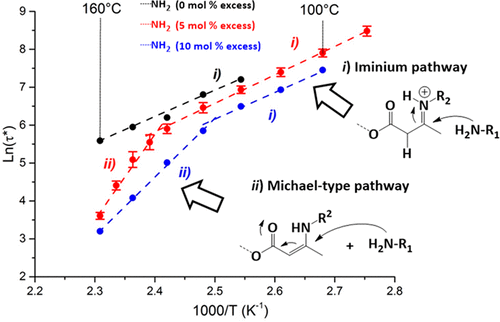
Vitrimers are an emerging new class of permanently cross-linked polymeric materials that show a liquid behavior upon heating wherein the macroscopic deformation is controlled by the rate of internal chemical bond exchange reactions. Thus, quite uniquely among polymeric materials, flow rates and material viscosities can be enhanced or controlled by the addition of catalysts and additives. We now report a catalyst-free vitrimer system, prepared from mixing two simple components, wherein two competing bond exchange mechanisms coexist, each showing a strikingly different temperature dependence, related to the large difference in activation energy for the different exchange pathways (60 vs 130-170 kJ/mol). The low barrier process is predominant at lower temperatures, but is outcompeted by the high barrier process that becomes dominant at higher temperatures because of its much more pronounced temperature dependence. The result is an interesting and highly unusual dual viscosity profile for this new class of vitrimer materials: a very gradual decrease in viscosity at lower temperatures, intercepted by a much sharper drop in viscosity at higher temperatures. The highly counterintuitive effect where a higher barrier pathway is dominant over a much lower barrier process can be rationalized by the exchange mechanisms that involve different reactive species, but lead to the overall same exchange. We observed this unusual but highly promising behavior first for fluorinated vitrimer elastomers, aimed at high performance materials, but the effect was also shown to hold in related nonfluorinated elastomers. A new way to control and design the rheological behavior of vitrimers toward finely tuned and precisely controlled processing applications has thus been provided.
Journal of the American Chemical Society, 140 (41), p13272-13284, 2018
https://pubs.acs.org/doi/10.1021/jacs.8b07094
36. Nonselective chemical inhibition of sec7 domain-containing arf gtpase exchange factors
Mishev K., Lu Q., Denoo B., Peurois F., Dejonghe W., Hullaert J., De Rycke R., Boeren S., Bretou M., De Munck S., Sharma I., Goodman K., Kalinowska K., Storme V., Nguyen L. S. L., Drozdzecki A., Martins S., Nerinckx W., Audenaert D., Vert G., Madder A., Otegui M. S., Isono E., Savvides S. N., Annaert W., De Vries S., Cherfils J., Winne J. and Russinova E.
Small GTP-binding proteins from the ADP-ribosylation factor (ARF) family are important regulators of vesicle formation and cellular trafficking in all eukaryotes. ARF activation is accomplished by a protein family of guanine nucleotide exchange factors (GEFs) that contain a conserved catalytic Sec7 domain. Here, we identified and characterized Secdin, a small-molecule inhibitor of Arabidopsis thaliana ARF-GEFs. Secdin application caused aberrant retention of plasma membrane (PM) proteins in late endosomal compartments, enhanced vacuolar degradation, impaired protein recycling, and delayed secretion and endocytosis. Combined treatments with Secdin and the known ARF-GEF inhibitor Brefeldin A (BFA) prevented the BFA-induced PM stabilization of the ARF-GEF GNOM, impaired its translocation from the Golgi to the trans-Golgi network/early endosomes, and led to the formation of hybrid endomembrane compartments reminiscent of those in ARF-GEF-deficient mutants. Drug affinity-responsive target stability assays revealed that Secdin, unlike BFA, targeted all examined Arabidopsis ARF-GEFs, but that the interaction was probably not mediated by the Sec7 domain because Secdin did not interfere with the Sec7 domain-mediated ARF activation. These results show that Secdin and BFA affect their protein targets through distinct mechanisms, in turn showing the usefulness of Secdin in studies in which ARF-GEF-dependent endomembrane transport cannot be manipulated with BFA.
Plant Cell, 30 (10), p2573-2593, 2018
http://www.plantcell.org/content/30/10/2573
35. Stereoselective and Modular Assembly Method for Heterocycle-Fused Daucane Sesquiterpenoids
Christiaens M., Hullaert J., Van Hecke K., Laplace D. and Winne J. M.
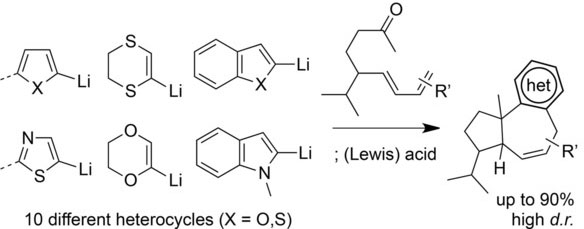
A stereoselective synthetic method is reported for the molecular framework found in common daucane and isodaucane sesquiterpenoid natural products. The synthetic method constitutes a scalable, modular, and also asymmetric access to a complex natural product scaffold, wherein the substitution pattern and the stereochemistry can be adjusted simply by choosing different starting materials. The method allows the rapid introduction of diverse heterocyclic substructures such as (benzo)furans, (benzo)thiophenes, dithiins, thiazoles, and indoles, which actually also facilitate and direct the key intramolecular annulation step.
Chemistry - A European Journal, 24 (52), p13783-13787, 2018
http://doi.wiley.com/10.1002/chem.201803248
34. Tunable Blocking Agents for Temperature-Controlled Triazolinedione-Based Cross-Linking Reactions
Houck H. A., De Bruycker K., Barner-Kowollik C., Winne J. M. and Du Prez F. E.
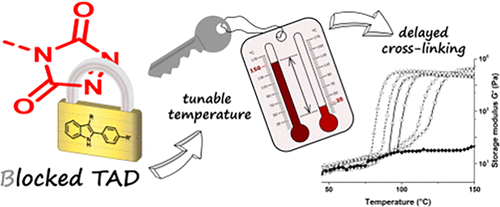
The remarkable reactivity of triazolinediones (TADs) toward olefin-type substrates marks them as highly attractive click reagents, in particular for polymer modification and cross-linking. Critically, their ultrafast reaction rates result in handling issues and a rather limited shelf life whereas a particular concern for polymer material applications is homogeneous network formation. Herein, we introduce 2-phenylindoles as highly promising blocking agents for TADs, giving bench-stable reagents at ambient temperature from which the initial TADs can be released upon heating. A set of 11 indoles with varying substitution patterns was synthesized, leading to a precise control for the temperature of deblocking within a broad range of 35-100 °C. The established indole-TAD platform was next applied to bivalent TAD reagents to enable on-demand TAD-based cross-linking reactions of a diene-containing polyurethane (Mn = 12.5 kDa). Fine-tuning of the curing temperatures, down to 50 °C, was evidenced via rheological measurements.
Macromolecules, 51 (8), p3156-3164, 2018
https://pubs.acs.org/doi/10.1021/acs.macromol.7b02526
33. Vinylogous Urea Vitrimers and Their Application in Fiber Reinforced Composites
Denissen W., De Baere I., Van Paepegem W., Leibler L., Winne J. M. and Du Prez F. E.
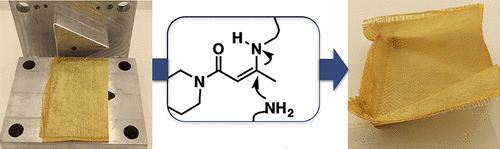
Vitrimers are covalently cross-linked polymeric materials that can be thermally processed in a liquid state without losing their network integrity. In this work, novel vitrimers based on the dynamic amine exchange reaction of vinylogous urea moieties are introduced. Following a systematic exploration of different vinylogous acyl compounds (urethanes, amides, and urea), vinylogous urea clearly emerges as showing the fastest intrinsic exchange kinetics. The combination of these networks with a simple acid catalyst (0.5 mol % pTsOH) resulted in materials with good mechanical properties (Tg ∼ 110 °C, E ∼ 2.2 GPa) and remarkably short relaxation times above Tg, in the order of a few seconds when heated. This attractive combination of properties made it possible to prepare vinylogous urea based composites as enduring prepregs. We demonstrate that the dynamic material properties give fully cured composites that still allow an efficient thermal fusion of multiple layers as well as thermoforming. Finally, we also demonstrate fiber recycling by a simple chemical treatment.
Macromolecules, 51 (5), p2054-2064, 2018
https://pubs.acs.org/doi/10.1021/acs.macromol.7b02407
2017
32. Polydimethylsiloxane quenchable vitrimers
Stukenbroeker T., Wang W., Winne J. M., Du Prez F. E., Nicolaÿ R. and Leibler L.
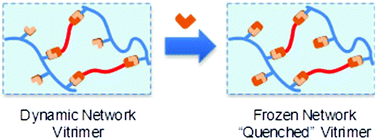
An elastomeric vitrimer material was synthesized via the crosslinking of a polydimethylsiloxane bearing pendant amino functions with a bis-vinylogous urethane crosslinker. The elastic properties of this material could be tuned across a wide range of moduli by varying crosslink density. Reshaping and recyclability were observed at 100 °C. Further investigation of the dynamic character revealed double relaxations, which could be supressed by masking free amino groups. Finally, these studies led to the development of a single-pot, solution-castable formulation of a non-dynamic permanent elastomer. These results open a way for the design of quenchable vitrimers, that is to say crosslinked materials which after shaping or recycling exhibit conventional elastomer properties.
Polymer Chemistry, 8 (43), p6590-6593, 2017
https://pubs.rsc.org/en/content/articlelanding/2017/PY/C7PY01488K
31. Heterocycles as Moderators of Allyl Cation Cycloaddition Reactivity
Hullaert J., Denoo B., Christiaens M., Callebaut B. and Winne J.
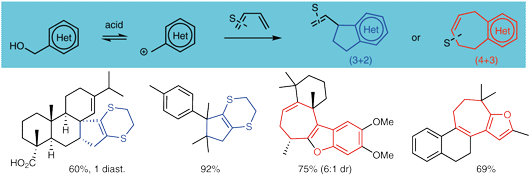
For the rapid elaboration of polycarbocyclic scaffolds, prevalent in many important families of terpenoid natural products, allyl cations derived from simple heterocyclic alcohols can be used as versatile reaction partners in both (4+3) and (3+2) cycloaddition pathways. Our recent progress in this area is outlined, pointing towards the untapped potential of heterocycles to act as reagents in novel or known but challenging organic transformations.
Synlett, 28 (18), p2345-2352, 2017
http://www.thieme-connect.de/DOI/DOI?10.1055/s-0036-1588511
30. Poly(thioether) Vitrimers via Transalkylation of Trialkylsulfonium Salts
Hendriks B., Waelkens J., Winne J. M. and Du Prez F. E.
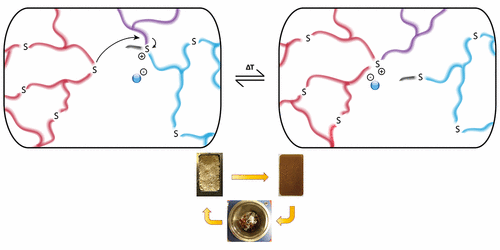
Vitrimers are permanently cross-linked organic polymers that can be reshaped, molded, and recycled without loss of network integrity. Herein, we report poly(thioether) networks, prepared through a straightforward thiol-ene photopolymerization, that can be turned into catalyst-free vitrimer materials by partial alkylation of the thioethers (1-10%) to the corresponding trialkylsulfonium salts. Based on a classical SN2-type substitution, the resulting polyionic networks can be reshaped upon heating via swift transalkylation reactions. This novel exchange reaction for the design of vitrimers was studied both on low MW model compounds as well as on a material level. In addition, we demonstrated the recycling of these networks without significant loss of mechanical properties.
ACS Macro Letters, 6 (9), p930-934, 2017
https://pubs.acs.org/doi/10.1021/acsmacrolett.7b00494
29. Covalent Fluorination Strategies for the Surface Modification of Polydienes
De Bruycker K., Delahaye M., Cools P., Winne J. and Prez F. E.
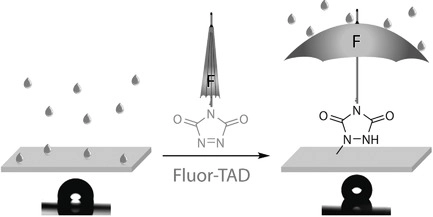
Nonreactive additives are widely applied to enhance polymer properties but can leach out of the material over time. In this work, two essentially different fluorinated additives bearing a triazolinedione moiety are synthesized and grafted on several polydiene backbones (acrylonitrile–butadiene–styrene, styrene–butadiene, and styrene–isoprene–styrene (SIS) copolymers), either by dip-coating or by reaction in solution. The resulting polymers are analyzed by contact angle goniometry, size exclusion chromatography, and NMR, infrared, and X-ray photoelectron spectroscopy. Independent of the modification procedure, the fluorophilic perfluoroalkyl additive is found at the material surface, thereby yielding a more hydrophobic surface. For SIS thermoplastic elastomers, for example, contact angles up to 125° can be obtained. (Figure presented.).
Macromolecular Rapid Communications, 38 (11), p1700122, 2017
http://doi.wiley.com/10.1002/marc.201700122
28. Micellar Paclitaxel-Initiated RAFT Polymer Conjugates with Acid-Sensitive Behavior
Louage B., van Steenbergen M. J., Nuhn L., Risseeuw M. D. P., Karalic I., Winne J. M., Van Calenbergh S., Hennink W. E. and De Geest B.
Acid-sensitive paclitaxel (PTX)–polymer conjugates were designed by applying a grafting-from-drug RAFT approach. PTX was linked through either a cyclic or a linear, acid-sensitive acetal moiety. Relative to direct esterification of PTX, which occurred regioselectively at the C2′ OH-group, direct acetalization was observed at either the C2′ or the C7 OH-group of PTX. This yielded two regioisomers of acetal-based PTX-functionalized RAFT chain transfer agents (CTAs). Subsequent polymerization with N,N-dimethylacrylamide (DMA) resulted in amphiphilic highly defined, acetal-based PTX–polymer conjugates with nearly identical features in terms of polymer definition and micellar self-assembly behavior, but with distinct PTX release kinetics and absence of burst release. This was further reflected by their in vitro biological performance, giving insights into the difference of the release mechanism between ester- and acetal-based PTX–polymer conjugates.
Macro Letters, 6 (3), p272-276, 2017
https://pubs.acs.org/doi/10.1021/acsmacrolett.6b00977
27. Chemical control of the viscoelastic properties of vinylogous urethane vitrimers
Denissen W., Droesbeke M., Nicola R., Leibler L., Winne J. M. and Du Prez F. E.
Vinylogous urethane based vitrimers are polymer networks that have the intrinsic property to undergo network rearrangements, stress relaxation and viscoelastic flow, mediated by rapid addition/elimination reactions of free chain end amines. Here we show that the covalent exchange kinetics significantly can be influenced by combination with various simple additives. As anticipated, the exchange reactions on network level can be further accelerated using either Br\onsted or Lewis acid additives. Remarkably, however, a strong inhibitory effect is observed when a base is added to the polymer matrix. These effects have been mechanistically rationalized, guided by low-molecular weight kinetic model experiments. Thus, vitrimer elastomer materials can be rationally designed to display a wide range of viscoelastic properties.
Nature Communications, 8 (1), p1-7, 2017
https://www.nature.com/articles/ncomms14857
26. Design of a thermally controlled sequence of triazolinedione-based click and transclick reactions
Houck H. A., De Bruycker K., Billiet S., Dhanis B., Goossens H., Catak S., Van Speybroeck V., Winne J. M. and Du Prez F. E.
The reaction of triazolinediones (TADs) and indoles is of particular interest for polymer chemistry applications, as it is a very fast and irreversible additive-free process at room temperature, but can be turned into a dynamic covalent bond forming process at elevated temperatures, giving a reliable bond exchange or 'transclick' reaction. In this paper, we report an in-depth study aimed at controlling the TAD-indole reversible click reactions through rational design of modified indole reaction partners. This has resulted in the identification of a novel class of easily accessible indole derivatives that give dynamic TAD-adduct formation at significantly lower temperatures. We further demonstrate that these new substrates can be used to design a directed cascade of click reactions of a functionalized TAD moiety from an initial indole reaction partner to a second indole, and finally to an irreversible reaction partner. This controlled sequence of click and transclick reactions of a single TAD reagent between three different substrates has been demonstrated both on small molecule and macromolecular level, and the factors that control the reversibility profiles have been rationalized and guided by mechanistic considerations supported by theoretical calculations.
Chemical Science, 8 (4), p3098-3108, 2017
https://pubs.rsc.org/en/content/articlelanding/2017/SC/C7SC00119C
25. Synthetic protocol for afcs: A biologically active fluorescent castasterone analog conjugated to an Alexa Fluor 647 dye
Winne J. M., Irani N. G., van den Begin J. and Madder A.
Synthetic derivatization of hormonally active brassinosteroids (BRs) can provide useful small molecule tools to probe BR signaling pathways, such as fluorescent analogs. However, most biologically active BRs are not suitable for direct chemical conjugation techniques because their derivatization typically requires extensive synthetic work and chemistry expertise. Here, we describe an operationally simple, two-step procedure to prepare and purify an Alexa Fluor 647-castasterone (AFCS) from commercially available materials. The reported strategy is also amenable to the introduction of various other amine-based labeling groups.
ACS Macro Letters, 1564 (3), p9-21, 2017
https://link.springer.com/protocol/10.1007%2F978-1-4939-6813-8_2
2016
24. Mitochondrial uncouplers inhibit clathrin-mediated endocytosis largely through cytoplasmic acidification
Dejonghe W., Kuenen S., Mylle E., Vasileva M., Keech O., Viotti C., Swerts J., Fendrych M., Ortiz-Morea F. A., Mishev K., Delang S., Scholl S., Zarza X., Heilmann M., Kourelis J., Kasprowicz J., Nguyen L. S. L., Drozdzecki A., Van Houtte I., Szatmári A., Majda M., Baisa G., Bednarek S. Y., Robert S., Audenaert D., Testerink C., Munnik T., Van Damme D., Heilmann I., Schumacher K., Winne J., Friml J., Verstreken P. and Russinova E.
Plant cells maintain strict proton gradients over different membranes. Here, Dejonghe et al. show that several protonophores, including the known tyrosine kinase inhibitor TyrphostinA23, inhibit clathrin-mediated endocytosis by disturbing these gradients and causing cytoplasmic acidification.
Nature Communications, 7 (1), p11710, 2016
http://www.nature.com/articles/ncomms11710
23. Vitrimers: permanent organic networks with glass-like fluidity
Denissen W., Winne J. M. and Du Prez F. E.
Vitrimers possess the unique property that they are malleable while being permanently cross-linked. This mini-review highlights the existing vitrimer systems in the period 2011–2015 with the main focus on their chemical origin.
Chemical Science, 7 (1), p30-38, 2016
http://xlink.rsc.org/?DOI=C5SC02223A
22. (5,6-Dihydro-1,4-dithiin-2-yl)methanol as a Versatile Allyl-Cation Equivalent in (3+2) Cycloaddition Reactions
Hullaert J. and Winne J. M.
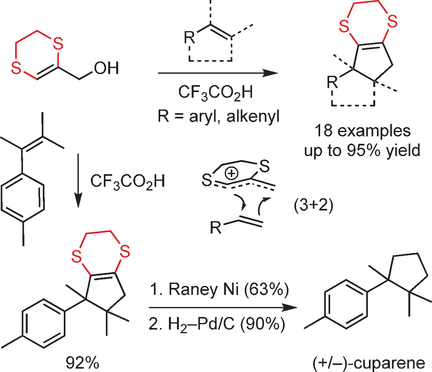
The title heterocyclic alcohol readily generates a sulfur-substituted allylic cation upon simple treatment with a protic acid, thus facilitating a synthetically useful stepwise (3+2) cycloaddition reaction pathway with a range of conjugated-olefin-type substrates. The introduction of an allyl fragment in this way provided rapid access to a variety of cyclopentanoid scaffolds. The cyclic nature of the cation precursor alcohol was shown to be instrumental for efficient cycloaddition reactions to take place, thus indicating an attractive strategy for controlling the reactivity of heteroatom-substituted allyl cations. The formal cycloaddition reaction is highly regio- and stereoselective and was also used for a short total synthesis of the natural product cuparene in racemic form through a cycloaddition-hydrodesulfurization sequence.
Angewandte Chemie International Edition, 55 (42), p13254-13258, 2016
http://doi.wiley.com/10.1002/anie.201606411
21. Triazolinediones as Highly Enabling Synthetic Tools
De Bruycker K., Billiet S., Houck H. A.,Chattopadhyay S., Winne J. M., Du Prez F. E.
Triazolinediones (TADs) are unique reagents in organic synthesis that have also found wide applications in different research disciplines, in spite of their somewhat "exotic" reputation. In this review, we offer two case studies that demonstrate the possibilities of these versatile and reliable synthetic tools, namely, in the field of polymer science as well as in more recently emerging applications in the field of click chemistry. As the general use of triazolinediones has always been hampered by the limited commercial and synthetic availability of such reagents, we also offer a review of the available TAD reagents, together with a detailed discussion of their synthesis and reactivity. This review thus aims to serve as a practical guide for researchers that are interested in exploiting and further developing the exceptional click-like reactivity of triazolinediones in various applications.
Chemical Reviews, 116 (6), p3919-3974, 2016
https://pubs.acs.org/doi/abs/10.1021/acs.chemrev.5b00599
2015
20. V-ATPase activity in the TGN/EE is required for exocytosis and recycling in Arabidopsis
Luo Y., Scholl S., Doering A., Zhang Y., Irani N. G., Di Rubbo S., Neumetzler L., Krishnamoorthy P., Van Houtte I., Mylle E., Bischoff V., Vernhettes S., Winne J., Friml J., Stierhof Y., Schumacher K., Persson S. and Russinova E.
The V-ATPase impaired in the det3 mutant acidifies trans-Golgi network/early endosome, and is needed for exocytosis of cargo vesicles, which are involved in recycling important membrane proteins such as the brassinoid receptor and cellulose synthase.
Nature Plants, 1 (7), p15094, 2015
http://www.nature.com/articles/nplants201594
19. From plant oils to plant foils: Straightforward functionalization and crosslinking of natural plant oils with triazolinediones
Türünç O., Billiet S., De Bruycker K., Ouardad S., Winne J. and Du Prez F. E.
With the aid of triazolinedione (TAD) chemistry, an additive-free, straightforward functionalization and crosslinking strategy for numerous plant oils was developed. In a first step, model studies on the most common natural fatty acids were performed with the aid of monofunctional TAD moieties. These equimolar functionalization reactions were readily monitored by NMR and MS analysis, further facilitated by the disappearance of the characteristic red colour of the TAD molecule. Then, a series of synthesized, bifunctional TAD molecules were used for the chemical crosslinking of crude plant oils, a process that was typically finished within minutes. In this way, a large variety of polymer networks could be obtained, such as plant oil-based foils, showing a wide range of thermal properties, which can be tuned and rationalized by the chemical structure of the different plant oils.
European Polymer Journal, 65, p286-297, 2015
https://www.sciencedirect.com/science/article/pii/S0014305714004431?via%3Dihub
18. Vinylogous Urethane Vitrimers
Denissen W., Rivero G., Nicolaÿ R., Leibler L., Winne J. M. and Du Prez F. E.
Vitrimers are a new class of polymeric materials with very attractive properties, since they can be reworked to any shape while being at the same time permanently cross-linked. As an alternative to the use of transesterification chemistry, we explore catalyst-free transamination of vinylogous urethanes as an exchange reaction for vitrimers. First, a kinetic study on model compounds reveals the occurrence of transamination of vinylogous urethanes in a good temperature window without side reactions. Next, poly(vinylogous urethane) networks with a storage modulus of ≈2.4 GPa and a glass transition temperature above 80 °C are prepared by bulk polymerization of cyclohexane dimethanol bisacetoacetate, m-xylylene diamine, and tris(2-aminoethyl)amine. The vitrimer nature of these networks is examined by solubility, stress-relaxation, and creep experiments. Relaxation times as short as 85 s at 170 °C are observed without making use of any catalyst. In addition, the networks are recyclable up to four times by consecutive grinding/compression molding cycles without significant mechanical or chemical degradation.
Advanced Functional Materials, 25 (16), p2451-2457, 2015
http://doi.wiley.com/10.1002/adfm.201404553
2014
17. A Rapid and Stereocontrolled Synthesis of the Zizaane Ring System by Using an Intramolecular (4+3) Cycloaddition Reaction
Laplace D. and Winne J.
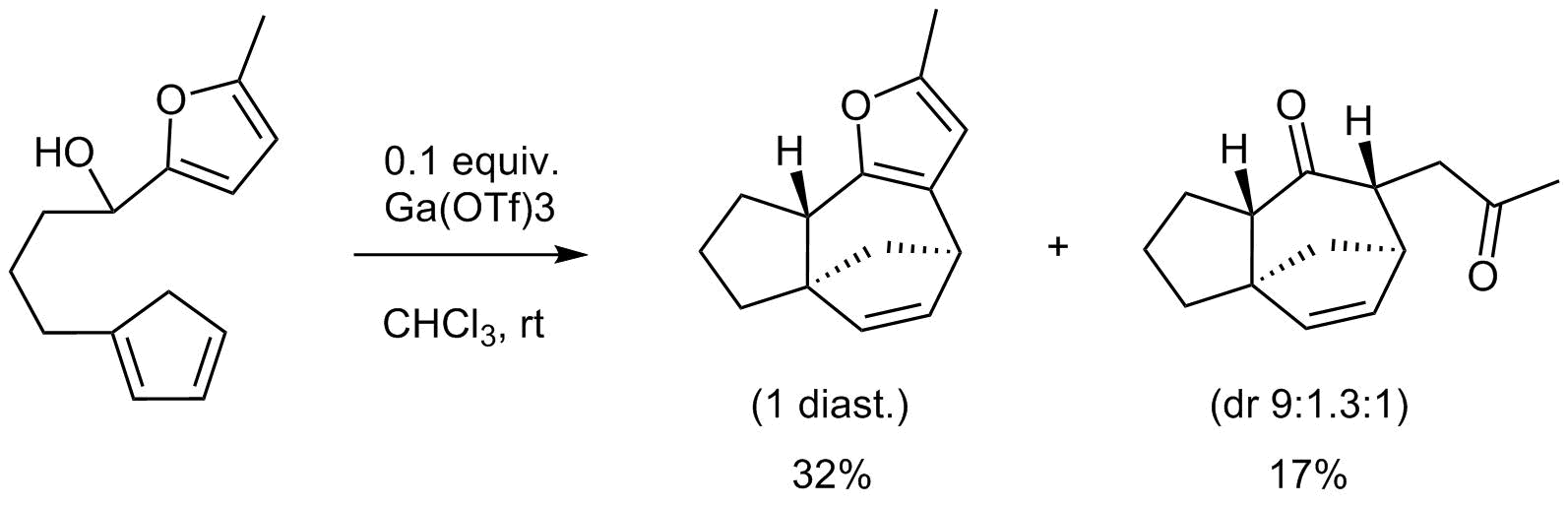
We report herein a very short synthetic approach to the tricyclo[6.2.1.01,5]undecane scaffold through a convergent and stereocontrolled route by using an intramolecular (4+3) cycloaddition of a simple furfuryl alcohol precursor with a pendant cyclopentadiene moiety. The formation of alternative reaction products under various conditions is also discussed.
Synlett, 26 (04), p467-470, 2014
http://www.thieme-connect.de/DOI/DOI?10.1055/s-0034-1378924
16. Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems
Billiet S., De Bruycker K., Driessen F., Goossens H., Van Speybroeck V., Winne J. M. and Du Prez F. E.
Macromolecular functionalization and linking are often facilitated by using 'click' chemistries. Now, triazolinediones have been used in ultrafast click reactions under additive-free, ambient conditions for polymer conjugation. Clicking to indoles gives an adduct that is dynamic at elevated temperatures, which produces properties such as polymer-network healing, reshaping and recycling.
Nature Chemistry, 6 (9), p815-821, 2014
http://www.nature.com/articles/nchem.2023
15. A Three-Step Synthesis of the Guaianolide Ring System
Hullaert J., Laplace D. R. and Winne J. M.
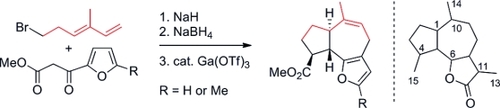
By using a gallium(III) triflate catalyzed intramolecular (4+3) cycloaddition, a few functionalized furan-derived tricycles that share the common guaianolide sesquiterpene ring system were prepared in a stereoselective manner in only three steps from commercially available starting materials. A discussion of the formation of alternative products is included, with possible substrate requirements to achieve the key cycloaddition step in an efficient way.
European Journal of Organic Chemistry, 2014 (15), p3097-3100, 2014
http://doi.wiley.com/10.1002/ejoc.201402170
14. Total Synthesis of (+/-)-Frondosin B and (+/-)-5- epi-Liphagal by Using a Concise (4+3) Cycloaddition Approach
Laplace D. R., Verbraeken B., Van Hecke K. and Winne J. M.
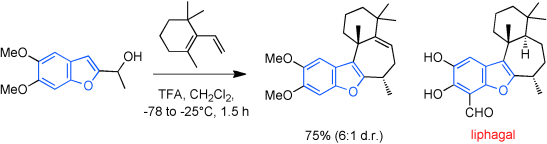
A recently developed (4+3) cycloaddition between dienes and furfuryl alcohols, as precursors of oxyallyl-type cations, has been used as a key step in the racemic syntheses of two natural products: frondosin B and liphagal. This work demonstrates the synthetic potential of this cycloaddition reaction, and offers a short synthetic route to an interesting family of natural products. A full account of these synthetic studies is presented, further illustrating the mechanism, scope, and limitations of this straightforward synthetic method for seven-membered rings.
Chemistry - A European Journal, 20 (1), p253-262, 2014
http://doi.wiley.com/10.1002/chem.201303273
2013
13. Synthesis of 2-Ethyl-19-nor Analogs of 1α,25-Dihydroxyvitamin D3
Laplace D. R., Van Overschelde M., De Clercq P. J., Verstuyf A. and Winne J. M.
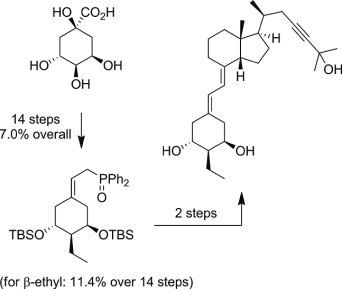
Two new phosphanone building blocks, useful for the stereocontrolled synthesis of 2-ethyl-substituted 19-nor analogs of 1α,25-Dihydroxyvitamin D3, were prepared from quinic acid. This route offers an attractive practical solution for the introduction of these synthetically challenging but highly interesting A-ring modifications.
European Journal of Organic Chemistry, 2013 (4), p728-735, 2013
http://doi.wiley.com/10.1002/ejoc.201201261
2011
12. Scope and Mechanism of the (4+3) Cycloaddition Reaction of Furfuryl Cations
Winne J. M., Catak S., Waroquier M. and Van Speybroeck V.
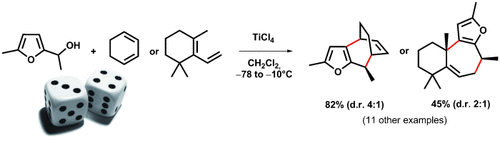
It all adds up: Furfuryl alcohols are revealed as direct reaction partners for a wide range of conjugated dienes in a (4+3) cycloaddition motif (see scheme). This novel Lewis acid promoted process gives straightforward access to various polycyclic skeletons containing a seven-membered ring. A plausible cationic stepwise mechanism was confirmed by DFT calculations.
Angewandte Chemie International Edition, 50 (50), p11990-11993, 2011
http://doi.wiley.com/10.1002/anie.201104930
Mentored work (PhD and postdoc)
2015
11. Possibility of [1,5] Sigmatropic Shifts in Bicyclo[4.2.0]octa-2,4-dienes
Goossens H., Winne J. M., Wouters S., Hermosilla L., De Clercq P. J., Waroquier M., Van Speybroeck V. and Catak S.
![Possibility of [1,5] Sigmatropic Shifts in Bicyclo[4.2.0]octa-2,4-dienes](images\publications\articles\article_23.gif)
The thermal equilibration of the methyl esters of endiandric acids D and E was subject to a computational study. An electrocyclic pathway via an electrocyclic ring opening followed by a ring flip and a subsequent electrocyclization proposed by Nicolaou [Nicolaou, K. C.; Chen, J. S. Chem. Soc. Rev. 2009, 38, 2993], was computationally explored. The free-energy barrier for this electrocyclic route was shown to be very close to the bicyclo[4.2.0]octa-2,4-diene reported by Huisgen [Huisgen, R.; Boche, G.; Dahmen, A.; Hechtl, W. Tetrahedron Lett. 1968, 5215]. Furthermore, the possibility of a [1,5] sigmatropic alkyl group shift of bicyclo[4.2.0]octa-2,4-diene systems at high temperatures was explored in a combined computational and experimental study. Calculated reaction barriers for an open-shell singlet biradical-mediated stepwise [1,5] sigmatropic alkyl group shift were shown to be comparable with the reaction barriers for the bicyclo[4.1.0]hepta-2,4-diene (norcaradiene) walk rearrangement.
The Journal of Organic Chemistry, 80 (5), p2609-2620, 2015
http://pubs.acs.org/doi/10.1021/jo5027639
2014
10. A strategy towards the synthesis of plumarellide based on biosynthesis speculation, featuring a transannular 4+2 type cyclisation from a cembranoid furanoxonium ion intermediate
Li Y., Palframan M. J., Pattenden G. and Winne J. M.
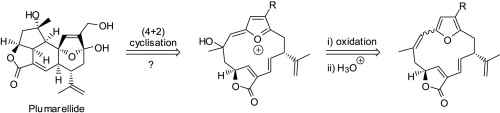
In studies towards a biomimetic synthesis of plumarellide 1, acid-catalysed rearrangement of the furanobutenolide-based acetonide 10b led to the ring system 15b in plumarellide together with the cycloheptene-based ring system 11 found in rameswaralide 4, in a combined yield of 60%. RCM of the ω-dienes 30a and 30b led to the macrocycles 38a and 38b, respectively with the Z-configuration. Treatment of the vinyl bromide/vinyl stannanes 32a and 47 under Stille conditions failed to give the macrocycles 31a and 48, respectively. Reactions of the macrocycles 38a and 38b with TFA containing water produced the 5,6,7-tricyclic compounds 56a and 56b, respectively in >90% yield, instead of the plumarellide-based ring system 52. A summary and perspective on possible pathways from the C-13 acetoxy furanocembranoid 66 and from the C13–C14 unsaturated enol ether/cyclic hemiketal 9/59 to plumarellide 1 in vivo is given.
Tetrahedron, 70 (40), p7229-7240, 2014
https://www.sciencedirect.com/science/article/pii/S0040402014009648?via%3Dihub
2013
9. Exploiting furan's versatile reactivity in reversible and irreversible orthogonal peptide labeling
Hoogewijs K., Buyst D., Winne J. M., Martins J. C. and Madder A.
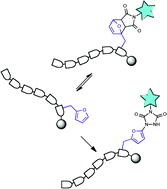
A general method for the facile and versatile decoration of peptides is proposed exploiting furan based cycloaddition and electrophilic aromatic substitution reactions. Given the commercial availability of furylalanine derivatives for peptide synthesis, the current work significantly enlarges the toolbox of available methodologies for site specific labeling and conjugation of peptide probes.
Chemical Communications, 49 (28), p2927, 2013
http://xlink.rsc.org/?DOI=c3cc40588e
2012
8. An Approach to exo-Enol Ether - Cyclic Ketal Structures Found in Marine Cembranoids, Based on Silver-Assisted Cyclisations of Enynone Precursors
Pattenden G. and Winne J.
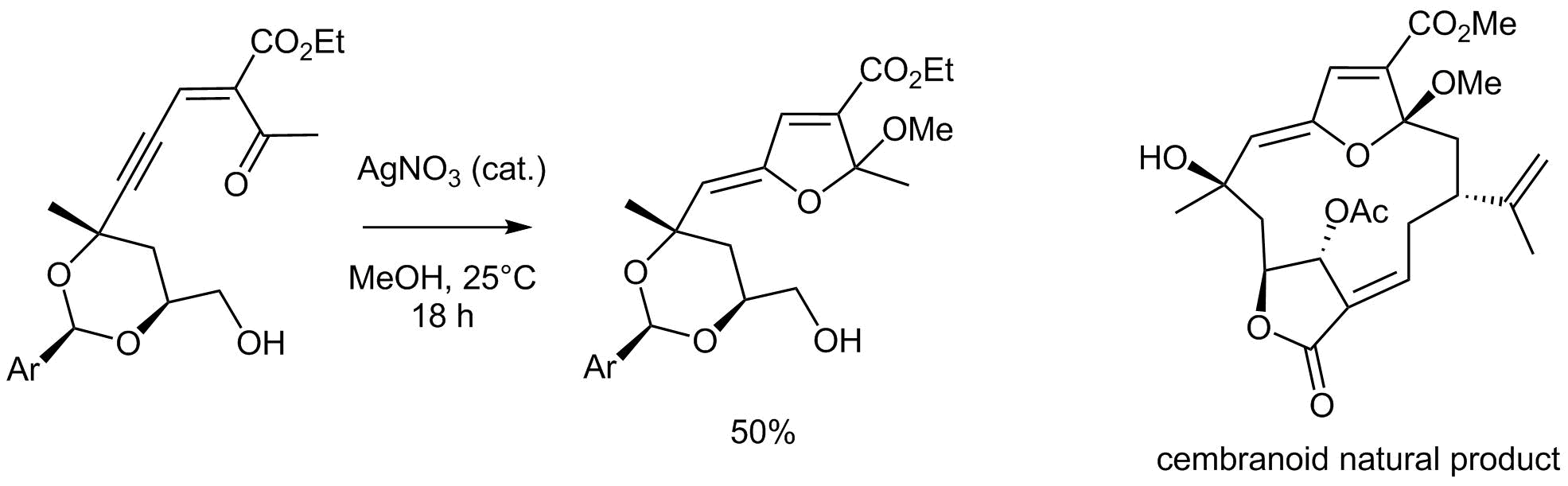
Treatment of a solution of the (E)-enynone 17 in methanol with AgNO3 leads to cyclisation and isolation of the substituted furan 25 in >95% yield, rather than the anticipated exo-enol ether-cyclic ketal structure 24. By contrast, a similar Ag-mediated cyclisation of the related substituted enynone 20 led to the exo-enol ether-cyclic ketal structure 28 (50%) alongside the substituted furan 29 (45%). The outcomes of these silver-assisted enynone cyclisations are compared and contrasted with earlier studies with acid-catalysed reactions of furanoepoxides, for example 7, which also lead to exo-enol ether-cyclic ketals, viz. 1, and to substituted furanmethanol products, that is, 10.
Synlett, 23 (5), p723-726, 2012
http://www.thieme-connect.de/DOI/DOI?10.1055/s-0031-1290363
2010
7. Synthetic studies towards oxygenated and unsaturated furanocembranoid macrocycles. Precursors to plumarellide, rameswaralide and mandapamates
Pattenden G. and Winne J. M.
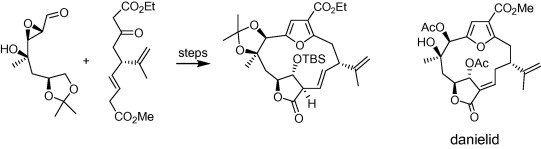
A new approach to the synthesis of oxygenated furanocembranoids, exemplified by danielid 3, is described. The approach features an intermolecular Knoevenagel-type condensation together with an intramolecular aldolisation to elaborate the furan and the lactone rings, respectively, in the initial target compound 14. Efforts to isomerise the C13–C14 double bond, or to introduce an alkene bond at C11–C12 in 14, leading to structures 3 and 8, respectively, are described.
Tetrahedron Letters, 51 (38), p5044-5047, 2010
https://www.sciencedirect.com/science/article/pii/S0040403910012955?via%3Dihub
2009
6. An intramolecular [4+3]-cycloaddition approach to rameswaralide inspired by biosynthesis speculation
Pattenden G. and Winne J. M.
![An intramolecular [4+3]-cycloaddition approach to rameswaralide inspired by biosynthesis speculation](images\publications\articles\Article_1.jpg)
A concise synthesis of the polycycle 27, which incorporates the 5,5,7-tricyclic ring core of rameswaralide 1, using a biogenetically inspired acid-catalysed [4+3]-cycloaddition approach starting from the furanobutenolide 26 is described, namely, 26→28/29→27. Under thermal conditions, the same furanobutenolide 26 gives the alternative polycyclic compound 35, resulting from a [4+2]-cycloaddition involving the furan as dienophile.
Tetrahedron Letters, 50 (52), p7310-7313, 2009
https://www.sciencedirect.com/science/article/pii/S0040403909019911?via%3Dihub
5. A synthetic approach to C-nor-D-homosteroids based on a cascade of radical cyclisations from a vinylcyclopropane-substituted acyl radical precursor
Pattenden G., Stoker D. A. and Winne J. M.
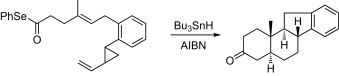
Treatment of the arylvinylcyclopropane-substituted seleno ester 5 with Bu3SnH–AIBN, under high dilution in benzene at 80°C, led to a 1:1 mixture of C10 methyl epimers of the C-nor-D-homosteroid ring system 24/25. The homosteroid was formed from 5 via a cascade of sequential acyl 13-endo trig radical macrocyclisation, benzyl radical 5-exo trig transannulation and alkyl radical transannulation reactions (Scheme 1). The macrocyclic dienone 23 was also isolated as an intermediate in the radical cascade between 5 and 24/25, and the dioxolanes 29 were interesting by-products. The cascade of radical cyclisations leading to the homosteroid 24/25 from the acyl radical precursor 5 is compared and contrasted with similar radical cascades from arylvinylcyclopropane-substituted alkyl radical precursors, i.e, 30→31 and 32b→38.
Tetrahedron, 65 (29-30), p5767-5775, 2009
https://www.sciencedirect.com/science/article/pii/S0040402009007169?via%3Dihub
4. A concise total synthesis of (±)-anthecularin
Li Y., Nawrat C. C., Pattenden G. and Winne J. M.
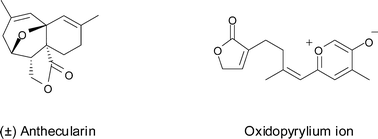
A total synthesis of the novel sesquiterpeneanthecularin 1, isolated from Greek Anthemis auriculata, based on an intramolecular [5+2] (1,3-dipolar) cycloaddition involving the oxidopyrylium ion 12 derived from the furanmethanol 9, is described.
Org. Biomol. Chem., 7 (4), p639-640, 2009
http://xlink.rsc.org/?DOI=B819454H
2008
3. Nonenzymic polycyclisation of analogues of oxidosqualene with a preformed C-ring
Winne J. M., De Clercq P. J., Milanesio M., Pattison P. and Viterbo D.
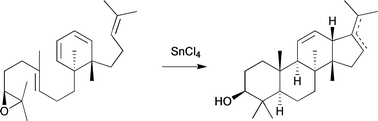
Some nonenzymic epoxide-initiated polyolefin cyclisations are reported. The presented molecules are partially constrained analogues of (3S)-oxidosqualene, the natural substrate to many important cyclase enzymes. These model compounds feature a preformed C-ring with built-in stereochemical information. The experimental results allow for an instructive comparison with the enzymic processes, particularly those of the cyclases in steroid biosynthesis (i.e. lanosterol synthase).
Organic & Biomolecular Chemistry, 6 (11), p1918, 2008
http://xlink.rsc.org/?DOI=b801670d
2006
2. Application of the B-Alkyl Suzuki-Miyaura Cross-Coupling Reaction to the Stereoselective Synthesis of Analogues of (3S)-Oxidosqualene
Winne J. M., Guang B., D'herde J. and De Clercq P. J.
A general method is described for the direct and stereoselective synthesis of epoxypolyenes via Suzuki-Miyaura cross-coupling reaction of 1-iodoalkenes with B-alkylboron compounds. It allows for the straightforward and convergent assembly of compounds that are structurally similar to (3S)-oxidosqualene, an important intermediate in steroid biosynthesis.
Organic Letters, 8 (21), p4815-4818, 2006